Диагностика во время беременности
Диагностика заболевания плода в период его вынашивания производится 2 способами: с помощью ультразвукового исследования, на котором выявляются грубые челюстно-лицевые нарушения, и молекулярного исследования образцов биологического материала.
Молекулярно-генетическая диагностика позволяет выявить изменения в ответственном гене путем прямого определения аминокислотной последовательности, которое производится автоматически.
В качестве биологического материала используются:
- образцы ворсин внешней оболочки зародыша, если срок беременности находится в пределах 8-14 недель;
- околоплодные воды, если женщина беременна на сроке 16-21 недель.
Стоимость исследования составляет порядка 30 тыс. руб., срок выполнения около 1 месяца, цена поиска мутации у родственника – 2800-3500 тыс. руб.
Если в семье уже имелись подобные случаи генетических нарушений, то настоятельно рекомендуется сделать диагностику перед подготовкой к беременности (стоимость исследования – 8-10 тыс. руб.). Такое исследование является единственным способом профилактики возникновения данной патологии у ребенка.
Диагностика у новорождённого
Определение заболевания у новорожденного обычно не вызывает затруднений и производится по внешним признакам. Генетический анализ для дополнительного подтверждения диагноза производится по венозной крови, таким же способом, как во время беременности.
Стоимость такого анализа порядка 60 тыс. руб. (вместе с заключением врача-генетика). При выявлении отклонения также рекомендуется сделать исследование для обоих родителей, братьев и сестер.
Дополнительно проводятся следующие обследования:
- определение остроты слуха, методика проведения зависит от возраста ребенка: регистрация вызванных слуховых потенциалов (подача акустических сигналов и регистрация электроэнцефалограммы), речевая аудиометрия (произнесение слов и фраз с различной громкостью), тональная пороговая аудиометрия (проводится с помощью аудиометров, результатом является графическое изображение слухового порога);
- рентгенография черепа;
- компьютерная томография височных костей по достижении возраста 3 лет для подготовки к хирургической операции;
- консультация у профильных специалистов (отоларинголога, окулиста, кардиолога, хирурга и других).
Данное заболевание требует дифференциальной диагностики с такими генетическими патологиями, как:
- Синдром Нагера. Для этой болезни также характерен антимонголоидный разрез глаз, недоразвитие нижней челюсти и патологические изменения в органах слуха и зрения. Кроме этого, у больных детей имеются грубые нарушения в развитии конечностей – укорочение и искривление предплечья, фаланг и локтевых костей, отсутствие пальцев и сращение костей рук.
- Синдром Гольденхара. Неправильное развитие затрагивает не только лицо и череп, но и нервную, сердечно-сосудистую, мочевыделительную, пищеварительную системы, у многих больных отмечается умственная отсталость. Как правило, данная аномалия имеет односторонний характер (70% больных).
Наиболее серьёзные осложнения
Помимо того что синдром и так ограничивает многие возможности человека, есть ещё и особые случаи, которые оказывают сильнейшее влияние на здоровье организма. Одним из наиболее тяжёлых осложнений принято считать деформацию ротового аппарата. Это связано с проблемами в строении зубов, что приводит к тому, что больной не может самостоятельно кушать.

Также могут возникнуть дополнительные сложности с дыханием, так как при синдроме Франческетти язык увеличивается в размерах и зарастают носовые проходы. Безусловно, эти факторы оказывают негативное влияние на дыхательную систему человека.
Стадии
Стадии синдрома определяются сложностью мутационного процесса и интенсивностью клинических признаков:
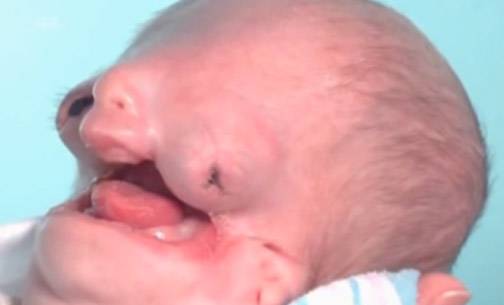
СТК тяжелой степени
- Начальная стадия характеризуется практически незаметными изменениями на лице. Больные дети ни чем не отличаются от здоровых и ведут нормальный образ жизни.
- Средняя стадия проявляется всеми перечисленными выше нарушениями. Аномальная деформация лицевых костей достаточно сильная. Возможны трудности с дыханием, принятием пищи, нарушение слуха, проблемы с зубами.
- Тяжелая стадия — полное отсутствие лица, невозможность рассмотреть его черты. Даже пластическая хирургия не может помочь больным.
Диагностика
Пренатальное исследование челюстно-лицевых аномалий проводится на 10-11 неделе беременности при помощи биопсии ворсин хориона. Процедура в достаточной степени опасная, поэтому в дородовой диагностике синдрома Тричера врачи предпочитают использовать УЗИ. Кроме того, у членов семьи берутся анализы крови. На 16-17 неделе беременности проводится процедура трансабдоминального амниоцентеза. Спустя некоторое время назначается фетоскопия и берется кровь из плодовых плацентарных сосудов.
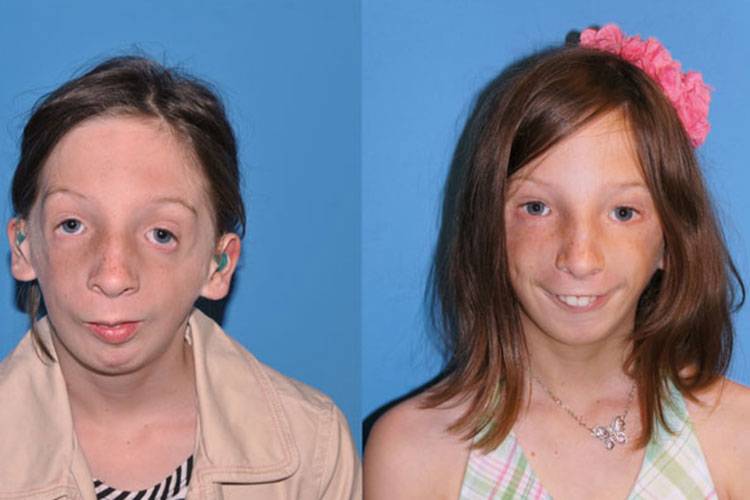
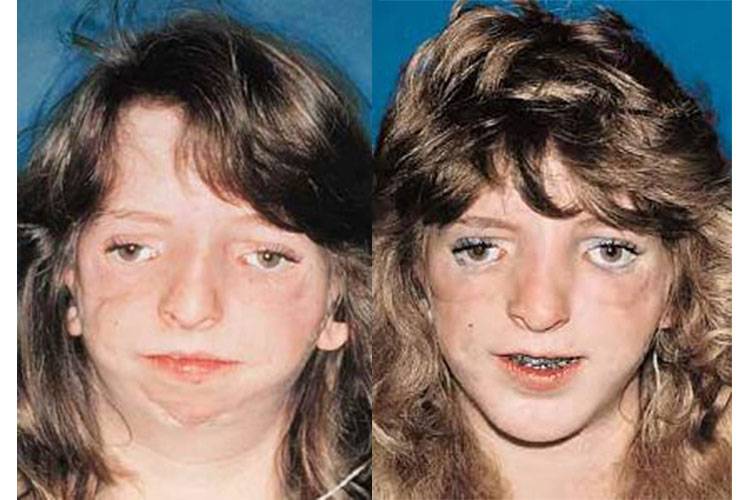

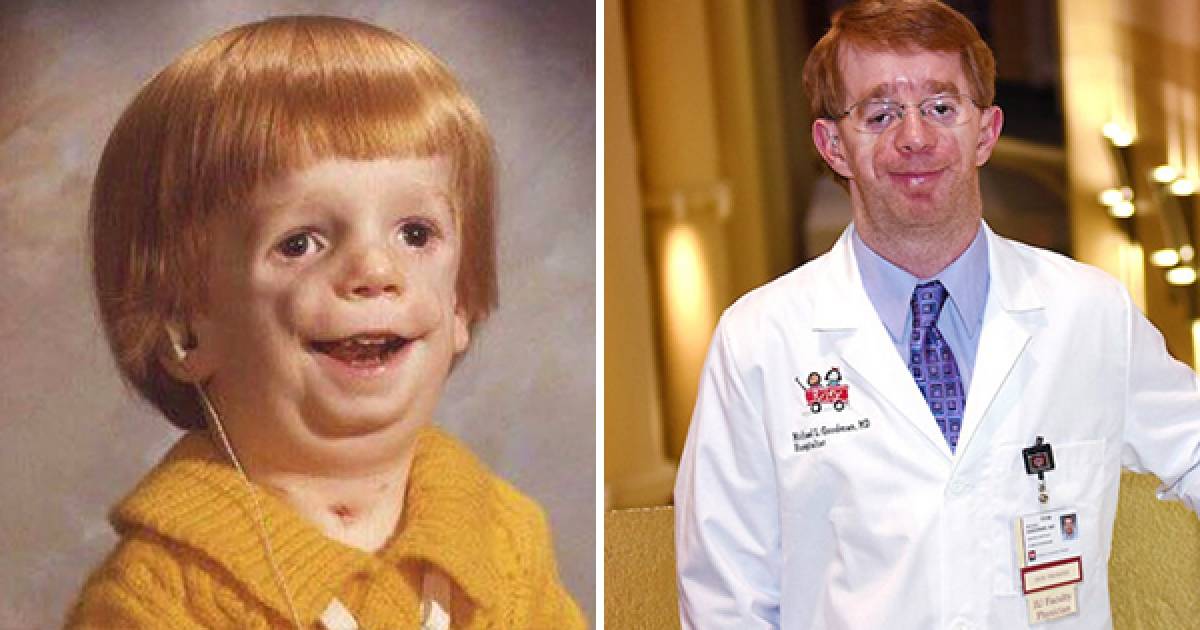
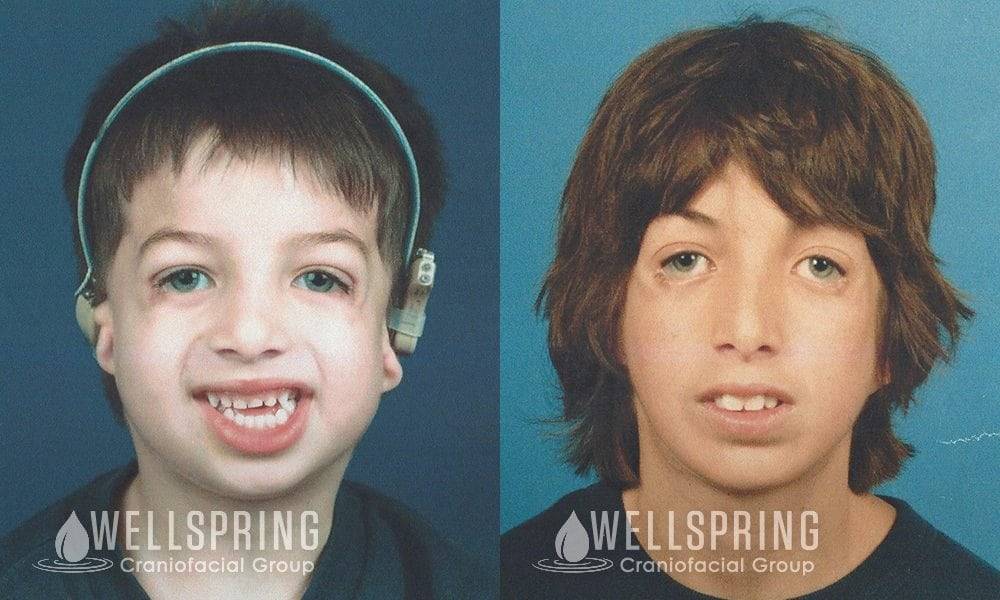
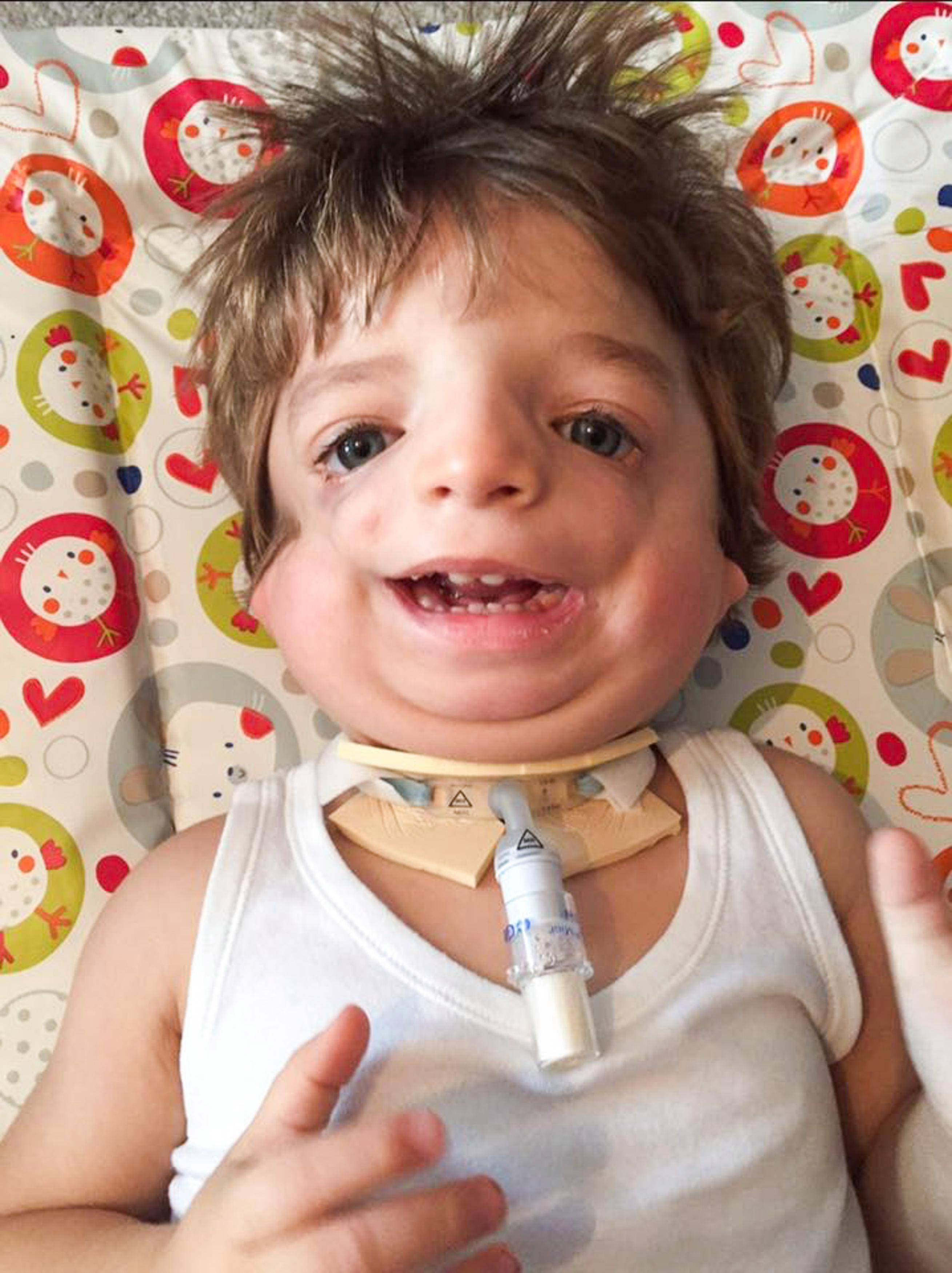

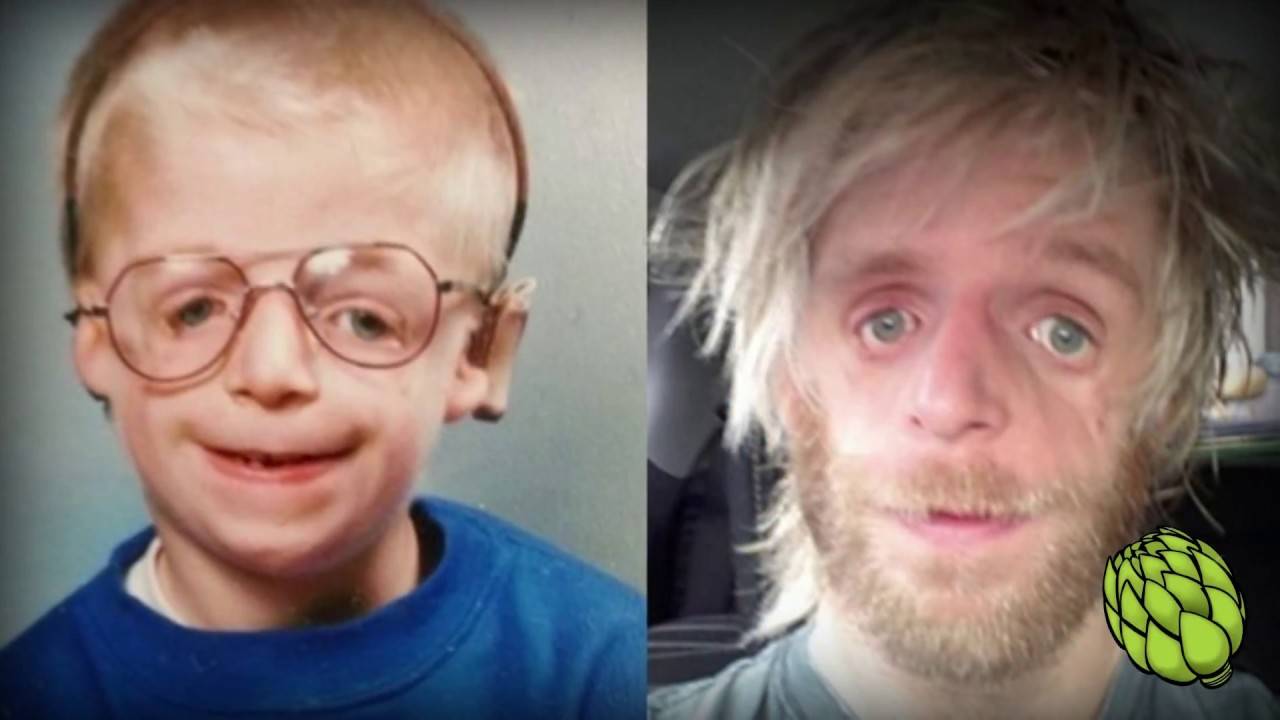
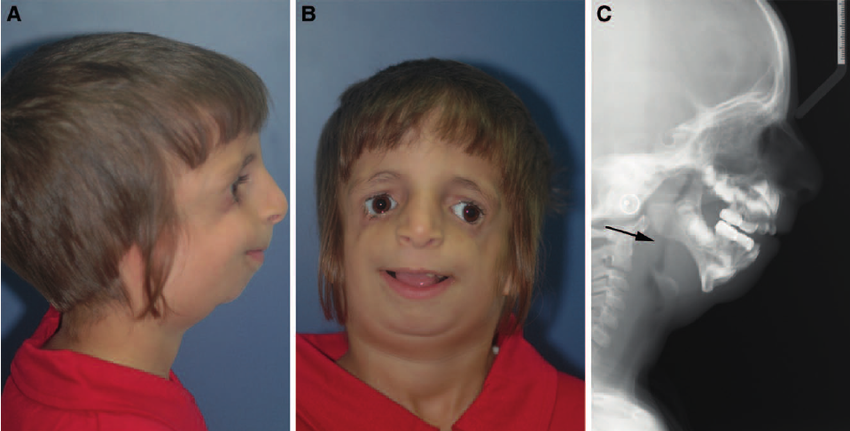
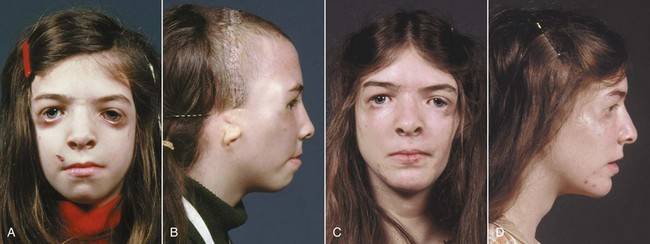
Постнатальная диагностика проводится на основании имеющихся клинических проявлений. При полной экспрессивности синдрома Тричера вопросов, как правило, не возникает, чего нельзя сказать, когда обнаруживаются незначительные признаки данной патологии. В этом случае проводится комплексная диагностика состояния, включающая следующие исследования:
- оценку и мониторинг эффективности кормления;
- аудиологическое тестирование слуха;
- рентгеноскопию черепно-лицевой дисморфологии;
- пантомографию;
- КТ или МРТ головного мозга.
Аналогичные методы исследования применяются, когда необходимо провести дифференциальную диагностику для того, чтобы распознать неярко выраженные проявления болезни Тричера-Коллинза и отличить их от признаков других патологических состояний. Так, в большинстве случаев специалисты назначают дополнительные инструментальные исследования для дифференциации указанного недуга с синдромами Гольденхара (гемифациальной микросомии), Нагера.
Последствия

Помимо внешних признаков патология себя не проявляет
Недуг очень серьезен и опасен своими осложнениями. Обычно это проблемы со слухом. Может быть недоразвит рот, что приводит к невозможному самостоятельному приему пищи. В результате зарастания носовых проходов могут происходить нарушения дыхательных функций. Также верхнее небо может быть недостаточно развитым и загромождать дыхательные ходы.
Помимо внешних признаков недуг больше никак себя не проявляет. Интеллектуальные способности больных детей абсолютно равны со здоровыми людьми. Такие пациенты полноценно развиваются, находят себя в жизни.
Многие нуждаются в квалифицированной психологической помощи. Уродства откладывают свой отпечаток на внутренних переживаниях личности, порождают множество комплексов, делают людей замкнутыми.
Такой диагноз носят пожизненно. Есть только один выход — сделать все возможные операции и жить полноценно, не обращая внимания на свои особенности.
Лечебные мероприятия
Как и для большинства генетических заболеваний, специального лечения при данном синдроме не существует. Терапия представляет собой сложную, многопрофильную задачу и зависит от тяжести поражения и осложнений.
Проводятся следующие лечебные мероприятия:
- В случае дыхательной недостаточности – трахеостомия. Эта процедура заключается в создании проходимости в трахее с помощью установки трубки для обеспечения поступления воздуха в дыхательные пути. В легких случаях возможна неинвазивная искусственная вентиляция легких.
- Дистракция (растяжение) нижней челюсти хирургическим путем с помощью специального аппарата. Необходимость проведения такой манипуляции рассматривается врачами в индивидуальном порядке.
- Установка гастростомы – трубки, которая требуется для защиты дыхательных путей от попадания пищи.
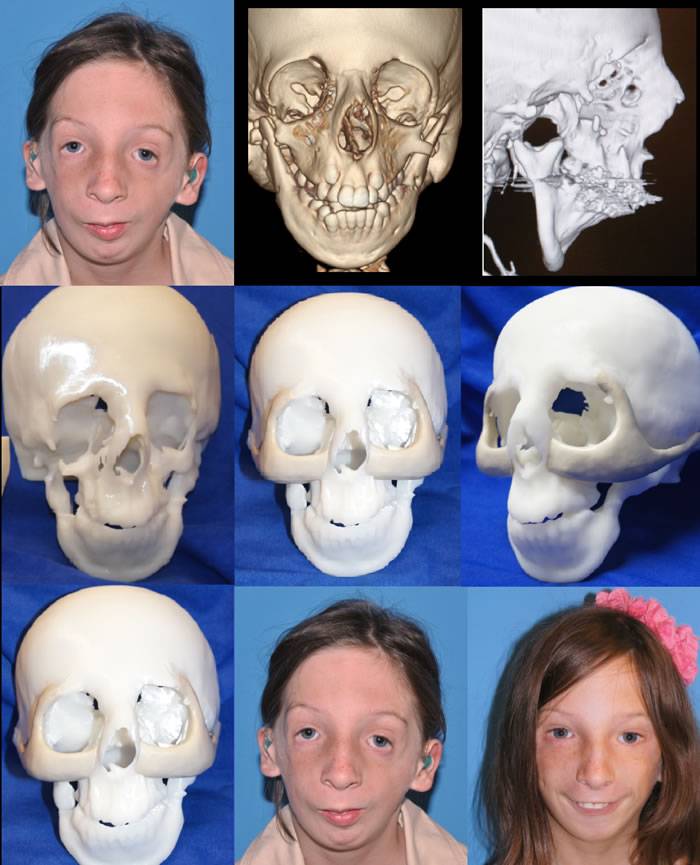
- Хирургическая реконструкция лицевой части черепа. Она обычно проводится с 5-6 лет.
При наличии осложнений показаны консультации у соответствующих специалистов и патогенетическое лечение.
Хирургические способы восстановления слуха у таких детей неэффективны. Рекомендуемая тактика реабилитации – использование слуховых аппаратов костной проводимости (или обычных слуховых аппаратов при незначительной деформации ушной раковины).
Их особенностью является то, что звуки передаются во внутреннее ухо по костям черепа. Это не так физиологично, как воздушное звукопроведение, однако при определенном усилении они очень хорошо воспринимаются рецепторами.
При данном заболевании хорошо зарекомендовали себя имплантируемые слуховые аппараты ВАНА (БАХА), которые имеют в своем составе титановую опору, закрепляемую в толще височной кости. Со временем титан срастается с костной тканью, а штифт напрямую передает звуковые колебания в улитку внутреннего уха.
Хирургическая установка имплантата производится в 2 этапа: сначала внедряется титановый штифт, а затем, после его вживления в кость в течение полугода, монтируется опора. Через месяц на нее надевают звуковой процессор.
Такие слуховые аппараты обладают следующими преимуществами:
- у детей отмечается улучшение порогов слышимости в диапазоне громкости обычной речи;
- по сравнению с обычными аппаратами и хирургической реконструкцией эстетические и аудиологические показатели выше;
- происходит спонтанное улучшение речи и постановки голоса (его высоты и интенсивности).
У маленьких детей слухопротезирование проводится с применением эластичной ленты, закрепляемой на голове. Существуют также цифровые аппараты костного проведения звуков, имплантируемые в височную кость (Alpha).


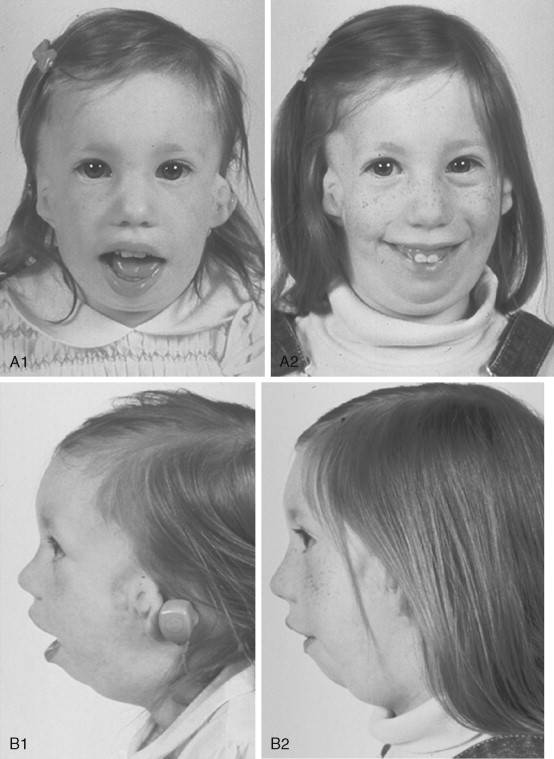
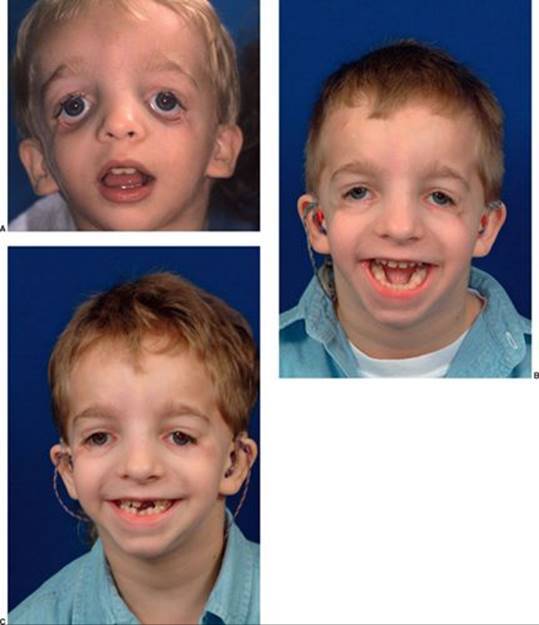



Лечение
Так как эта болезнь носит генетический характер, то лечения её просто не существует. Однако при сильно выраженных пороках развития лица есть возможность провести операцию, которая поможет устранить имеющиеся дефекты.
При необходимости проводится коррекция ушных раковин, волчьей пасти и пластика наружного слухового прохода. Так как при этом заболевании челюсти очень маленькие, а язык имеет большие размеры, то он просто не может поместиться во рту. Чтобы побороть эту патологию, проводится операция по удалению надгортанника и установке постоянной трахеостомы.
Вылечить этот серьёзный недостаток за одну операцию просто невозможно, особенно если нарушения выявлены в тяжёлой степени. Поэтому требуется несколько пластических операций, а сам цикл лечения может растянуться на несколько лет. Однако иногда удалить все дефекты не получается, и человеку приходится мириться с этим всю жизнь.
Причины заболевания
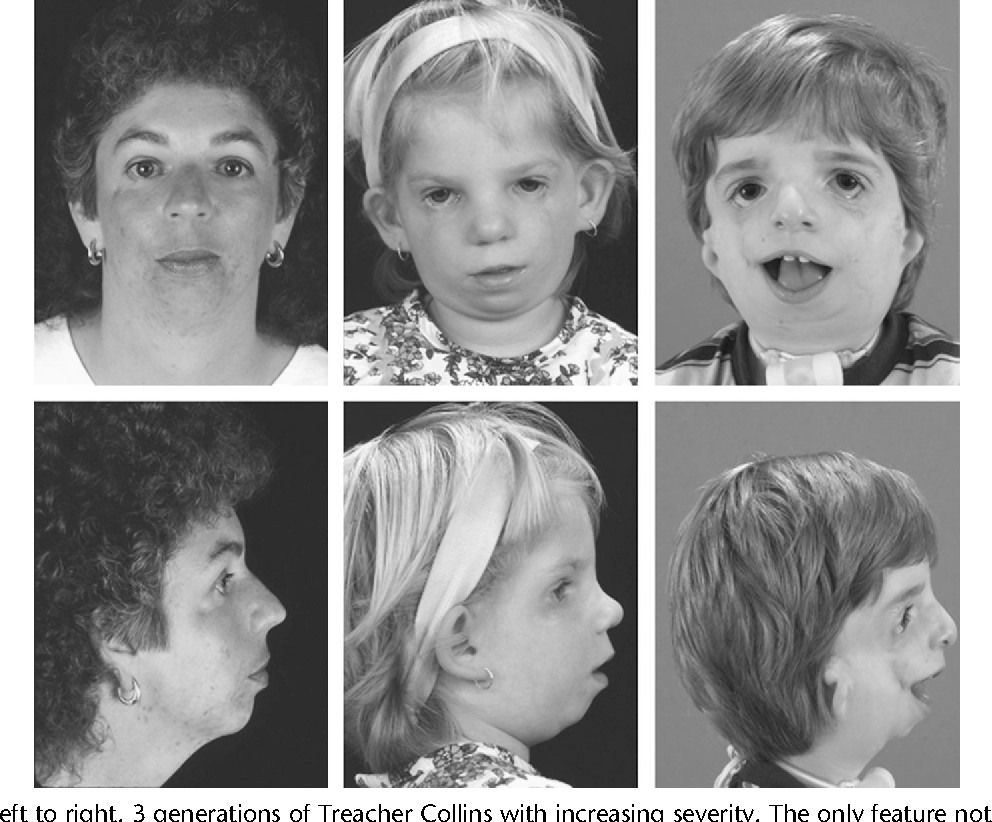
Синдром Тричера – генетическое заболевание, на возникновение которого в большинстве случаев не влияют никакие внешние или внутренние факторы. Можно сказать, что патология изначально заложена в аминокислотный код будущего ребенка и начинает проявляться задолго до его рождения. Научно доказано, что спонтанные изменения в структуре ДНК (генные мутации) у лиц, имеющих синдром, возникают в 5 хромосоме. Последняя является самой длинной нуклеотидной структурой в геноме человека и отвечает за производство материала для скелета плода.
Происходят мутации по причине сбоя внутриклеточного синтеза белка. В результате чего развивается синдром гаплонедостаточности. Последний характеризуется нехваткой белка, необходимого для правильного развития лицевой части черепа. При всем этом следует знать, что болезнь Тричера-Коллинза имеет аутосомно-доминантный, реже – аутосомно-рецессивный характер. Дефект генов наследуется детьми от больных родителей только в 40% случаев, тогда как остальное 60% возникают вследствие новых мутаций, которые нередко вызывают следующие тератогенные факторы:
- этанол и его производные;
- цитомегаловирус;
- радиоактивное излучение;
- токсоплазмоз;
- прием противосудорожных и психотропных препаратов, лекарств с ретиноевой кислотой.
Какие гены связаны с этим синдромом?
Мутации в гене TCOF1, POLR1C или POLR1D могут вызывать Treacher Collins. Мутации гена TCOF1 являются наиболее распространенной причиной расстройства, составляя от 81 до 93 процентов всех случаев. Мутации гена POLR1C и POLR1D вызывают дополнительные 2 процента случаев. У лиц без идентифицированной мутации в одном из этих генов генетическая причина заболевания неизвестна.
Белки, полученные из генов TCOF1, POLR1C и POLR1D, как представляется, играют важную роль в раннем развитии костей и других тканей лица. Эти белки участвуют в получении молекулы, называемой рибосомальной РНК (рРНК), химической кузены ДНК. Рибосомальная РНК помогает собрать белковые строительные блоки (аминокислоты) в новые белки, которые необходимы для нормального функционирования и выживания клеток. Мутации в гене TCOF1, POLR1C или POLR1D уменьшают продукцию рРНК. Исследователи предполагают, что уменьшение количества рРНК может спровоцировать саморазрушение (апоптоз) определенных клеток, участвующих в развитии костей и тканей лица. Аномальная гибель клеток может привести к специфическим проблемам с развитием лица, обнаруженным в TCS. Однако неясно, почему эффекты снижения рРНК ограничиваются развитием лица.
В настоящее время нет лечения для TCS. Лечение адаптировано к конкретным потребностям каждого ребенка или взрослого. В идеале лечение управляется многопрофильной командой специалистов-черепно-лицевых.
Новорожденным может потребоваться специальное позиционирование или трахеостомия для управления дыхательными путями. Потеря слуха может рассматриваться как усиление костной проводимости, речевая терапия и / или образовательная интервенция.
Во многих случаях необходима черепно-лицевая реконструкция. Хирургия может быть выполнена для восстановления расщепления неба, для восстановления челюсти или для восстановления других костей в черепе. Конкретные хирургические процедуры и возраст при проведении операции зависят от тяжести аномалий, общего состояния здоровья и личных предпочтений.
Есть несколько возможных методов лечения, которые исследуются. Исследователи ищут способы ингибировать протеин p53, который помогает организму убивать нежелательные клетки. У людей с TCS p53 аномально активируется, что приводит к потере определенных клеток и, в конечном счете, вызывает функции TCS. Было высказано предположение, что ингибирование продуцирования р53 (или блокирование его активации) может помочь лечить пораженных людей. Тем не менее, необходимы дополнительные исследования, чтобы определить, эффективен ли этот тип лечения и безопасен ли он.
Исследователи также изучают использование клеток стеблей, обнаруженных в жировой ткани, которые будут использоваться вместе с операцией у людей с ТКС и другими черепно-лицевыми расстройствами. Ранние исследования показали, что хирургические результаты могут быть улучшены с использованием этих стволовых клеток, чтобы помочь стимулировать восстановление роста пораженных участков. Однако эта терапия все еще экспериментальна и противоречива.
Синдром Тричера Коллинза причины
Синдром Тричера Коллинза является аутосомно — доминантным заболеванием. Это значит, что дефектный ген этой болезни не связан с половой хромосомой, а значит, равносильно может появиться и у мужчин и у женщин. Кроме того, этот ген является доминантным, что означает, что при его наличии в организме, синдром Тричера Коллинза проявится в 100% случаев наличия этого гена.
Таким образом, развитие заболевания Тричера Коллинза не зависит от воздействия каких-либо вредных внешних и внутренних факторов. Можно сказать, что это заболевание уже заложено в генетический код будущего ребенка и начинает раскрываться задолго до его рождения.
Причина появления синдрома Тричера Коллинза у новорожденного ребенка кроется еще на этапе эмбриогенеза плода и процесса закладки органов. Именно в это время, а точнее на 7-й неделе развития эмбриона, происходит мутация в определенной хромосоме человеческого генетического кода – 5-й хромосоме. Эта хромосома является самой длинной в геноме человека, и именно она отвечает за синтез материала для будущего костного скелета. В результате в этой хромосоме происходит особая мутация — так называемая «нонсенс-мутация». Особенность этой мутации кроется в особенностях внутриклеточного синтеза белка. В норме такой важный процесс как биосинтез белка происходит следующим образом: цепь ДНК «перезаписывает» информацию на вспомогательную единицу – РНК. Буквально говоря РНК клонируется, записывая на себя определенную последовательность участков ДНК. Эти участки представляют собой определенную последовательность составных частей нуклеотидов, каждый из которых несет свою особую информацию. Но кроме нуклеотидов существуют и особые гены, которые получили название «стоп-кодонов». Эти гены выполняют особую функцию — в последующей сборке белка на основе РНК они в заканчивают постройку молекулы белка.
Народ опешил! Суставы восстановятся за 3 дня! Приложите…
Мало кто знает, но именно это лечит суставы за 7 дней!





После того, как будет создана РНК с полной информацией, аналогичной информации материнской ДНК, она транспортируется в особый клеточный орган-рибосому. Именно она занимается синтезированием будущего белка — основы клеточной структуры определенных органов. РНК проходит сквозь рибосому, а точнее через ее функциональные участки. Эти участки взаимодействуют с РНК, «считывают» с нее информацию и вырабатывают белковые цепи, где каждая из них соответствует своему нуклеотиду на РНК. Но когда функциональный центр взаимодействует с описанным выше геном стоп кодоном, то он получает информацию о прекращении синтеза белка. Говоря своими словами, стоп кодоны, как бы «отрезают» от общей массы отдельные полипептидные цепи, где каждая имеет свою структуру. Позже эти цепи будут собраны в белковые молекулы.
Но при синдроме Тричера Коллинза в 5-й хромосоме, в ее гене под названием TCOF1 происходит сбой — на месте нормальных нуклеотидов, способных создать полипептидную цепь, образуется стоп-кодон. В результате, при дальнейшем синтезе происходит преждевременное окончание сборки белка, и такой белок получается дефектным. Как результат, развивается синдром гаплонедостаточности — количества образуемого белка при такой недостаточности просто недостаточно для синтезирования будущего прототипа костной структуры лицевой части черепа и последующего правильного развития из него самой костной структуры.
Как следствие этого, развивается формирование целого ряда деформаций лицевой части черепа: нарушение пропорций ее костной части, атрезия (недоразвитие) ушных раковин и наружного слухового прохода, полное или частичное нарушение формирования правильных черт лица.
Лечение
В случае, когда заболевание диагностировано еще во время внутриутробного развития, женщине рекомендуют сделать аборт. В противном случае, сразу после рождения ребенку потребуется квалифицированное комплексное лечение по устранению дефектов, насколько это возможно. Терапия заключается в следующих действиях:
- Хирургическое вмешательство, в ходе которого устраняются дефекты лица. Обычно необходимо несколько операций, чтобы уменьшить проявления болезни. Все зависит от сложности случая.
- Стоматологические процедуры. Обычно у ребенка диагностируют не только проблемы с зубами, но и не правильный прикус, которые с помощью дантистов можно корректировать.
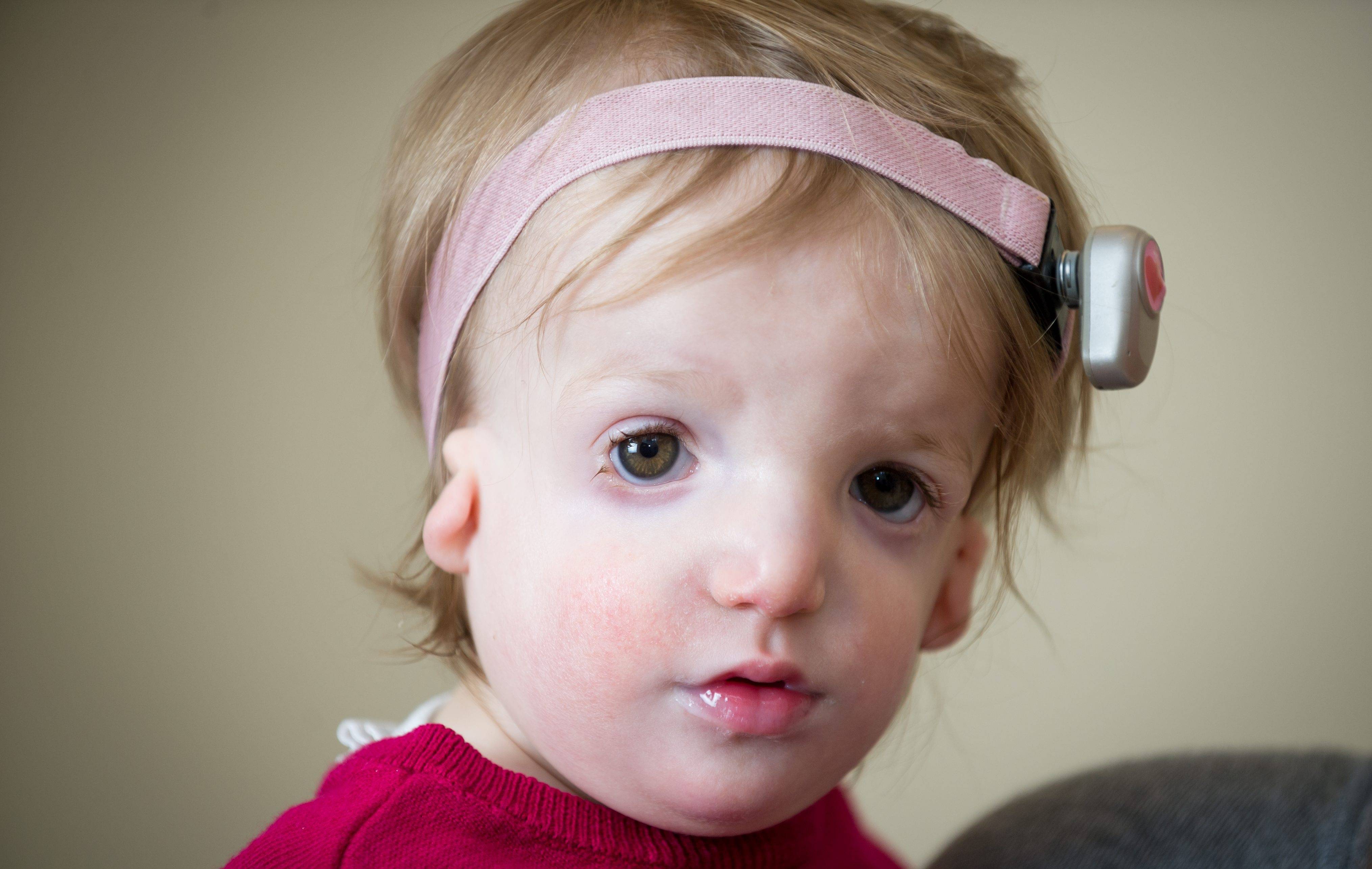
- Чтобы улучшить костнопроводимость звуков, потребуется установить слуховые аппараты.
Если у ребенка наблюдаются проблемы с дыханием и питанием, то потребуются операции по решению этих проблем (трахеотомия и гастростомия). Для дальнейшей адаптации ребенка потребуется работа с логопедами и сурдопедагогами.
Что это такое?
Как уже было отмечено, синдром Коллинза является исключительно генетическим заболеванием. Его невозможно приобрести в течение жизни, он проявляется с рождения. Данная болезнь – один из подвидов дизостоза, который, в свою очередь, означает нарушение или неправильное развитие костных тканей. Если говорить о синдроме Франческетти, то здесь происходит деформация костей черепа, что не может не оказывать влияние на физическое и моральное состояние человека.

У новорождённого сразу заложены клинические проявления, от количества которых зависит тяжесть развития заболевания. Стоит отметить, что болезнь характеризуется нарушением в строении черепа, которое отображается на лице человека. Чаще всего страдают от синдрома дети, у родителей которых наблюдаются спонтанные мутации генов.
Возможные осложнения и прогноз
Если заболевание не сопровождается грубыми пороками развития внутренних органов, то прогноз для жизни и здоровья ребенка в будущем благоприятен. При наличии тугоухости снижается способность к развитию речи, письма, обучению.
Необычная внешность также мешает получению навыков социального общения, так как другие дети стараются избегать сверстников с врожденными аномалиями и уродствами. В результате у ребенка может быть значительно снижена самооценка, что требует консультации со стороны психолога. Проведение ряда пластических операций позволяет частично сгладить дефекты развития.
Тугоухость часто ошибочно диагностируется как недоразвитие психики. Умственная отсталость у таких детей встречается редко, а некоторые имеют способности выше среднего
Поэтому у таких пациентов важно на ранней стадии выявлять нарушения слуха и корректировать их
Врачи рекомендуют это делать в обязательном порядке до достижения ребенком шестимесячного возраста. С 3-х месяцев он может носить аппарат для костного проведения звуковых волн, а после 3 лет возможна установка имплантата за ушами. Также очень большое значение имеют занятия с сурдопедагогом.
В первые месяцы жизни недоразвитость нижней челюсти может способствовать выпадению языка и перекрытию дыхательных путей, что является потенциально опасным для жизни ребенка. В последующем могут возникнуть затруднения в приеме пищи из-за ограниченной возможности открытия рта (в различной степени тяжести).
Диагностика
Необходимо понимать, что диагностировать генное заболевание можно уже на первых порах беременности. Для того чтобы увидеть, есть ли какие-то отклонения во внутриутробном развитии плода, проводят УЗИ. На основании результатов можно увидеть какие-либо изменения или нарушения со стороны развития костно-лицевой системы. Чтобы подтвердить диагноз рекомендуют провести генетический тест, который сможет либо подтвердить, либо опровергнуть подозрения.
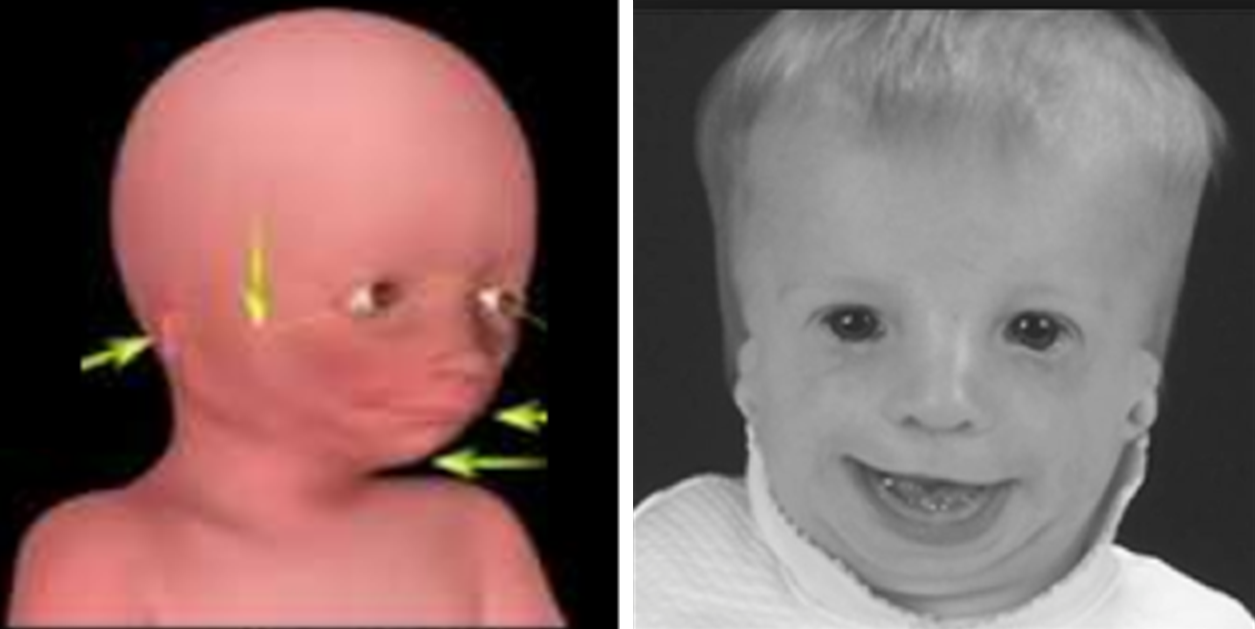
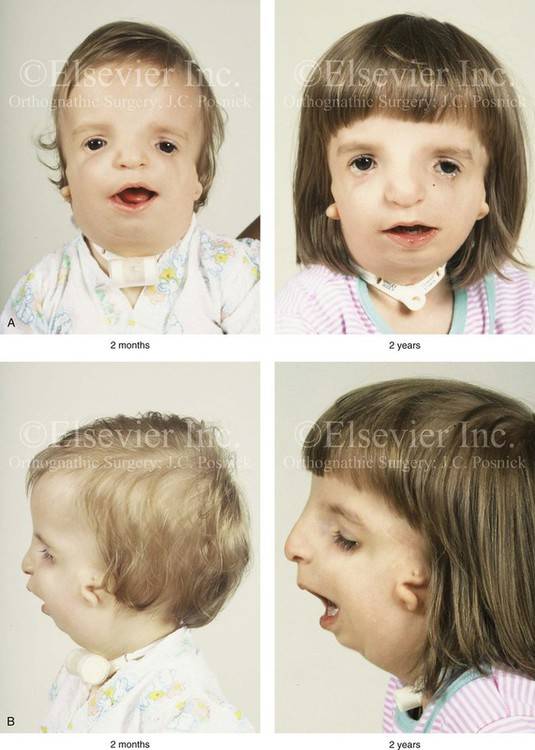

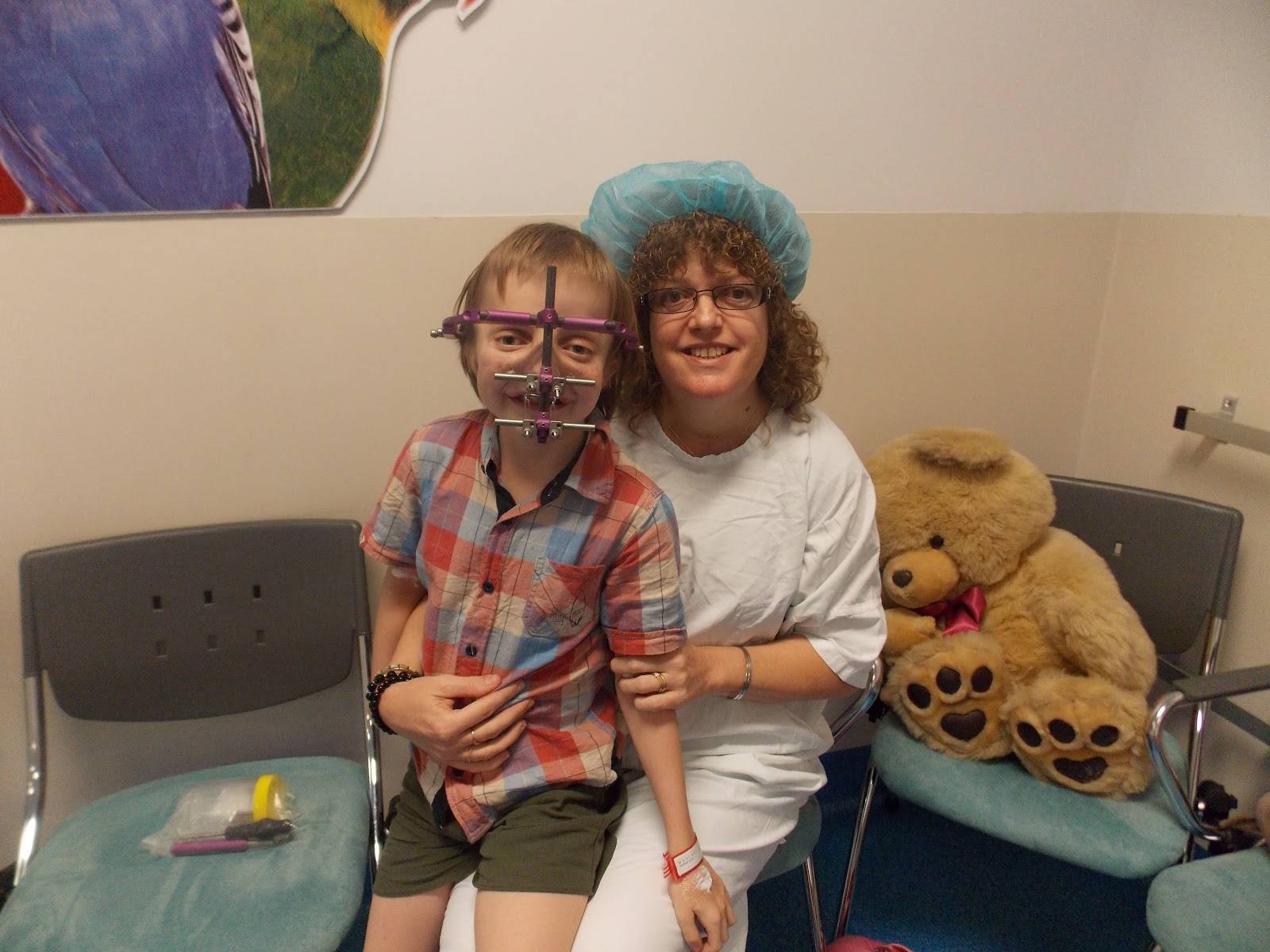


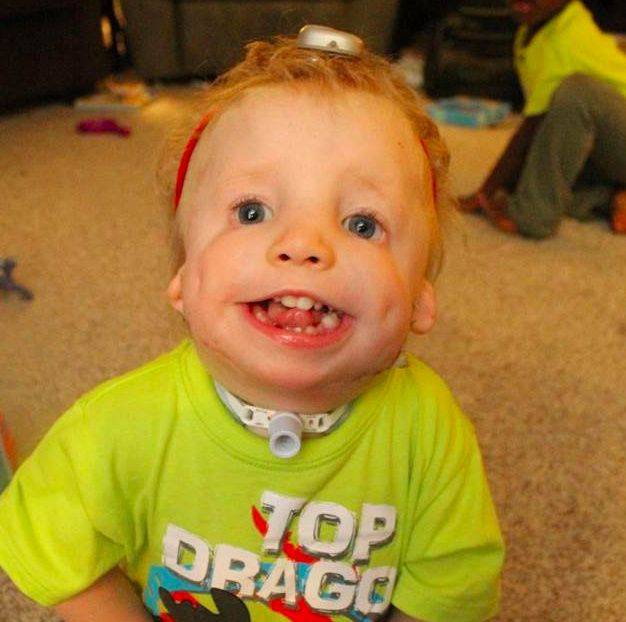


Кроме того, после родов также проводится визуальный осмотр ребенка, а в дальнейшем проверяется его слух. Основные методы оценивания выбираются от возраста:
- Регистрация слуховых потенциалов;
- Тональная и речевая аудиометрия.
Для выявления аномального развития слухового аппарата проводится томография височной части.
Диагностика
Заподозрить формирование недуга можно еще на этапе эмбрионального развития. Характерные изменения лицевых костей черепа видны на УЗИ. Для подтверждения наличия генетического заболевания потребуется проведение кариотипирования. Неонатальная диагностика синдрома Франческетти-Коллинза основана на специфических клинических признаках. Важным этапом обследования является оценка работы слухового анализатора. В ряде случаев для исключения других возможных проблем проводится клиническое и биохимическое исследование крови, УЗИ, а также магнитно-резонансная и компьютерная томография. Окончательный диагноз ставится на основании генетических тестов.
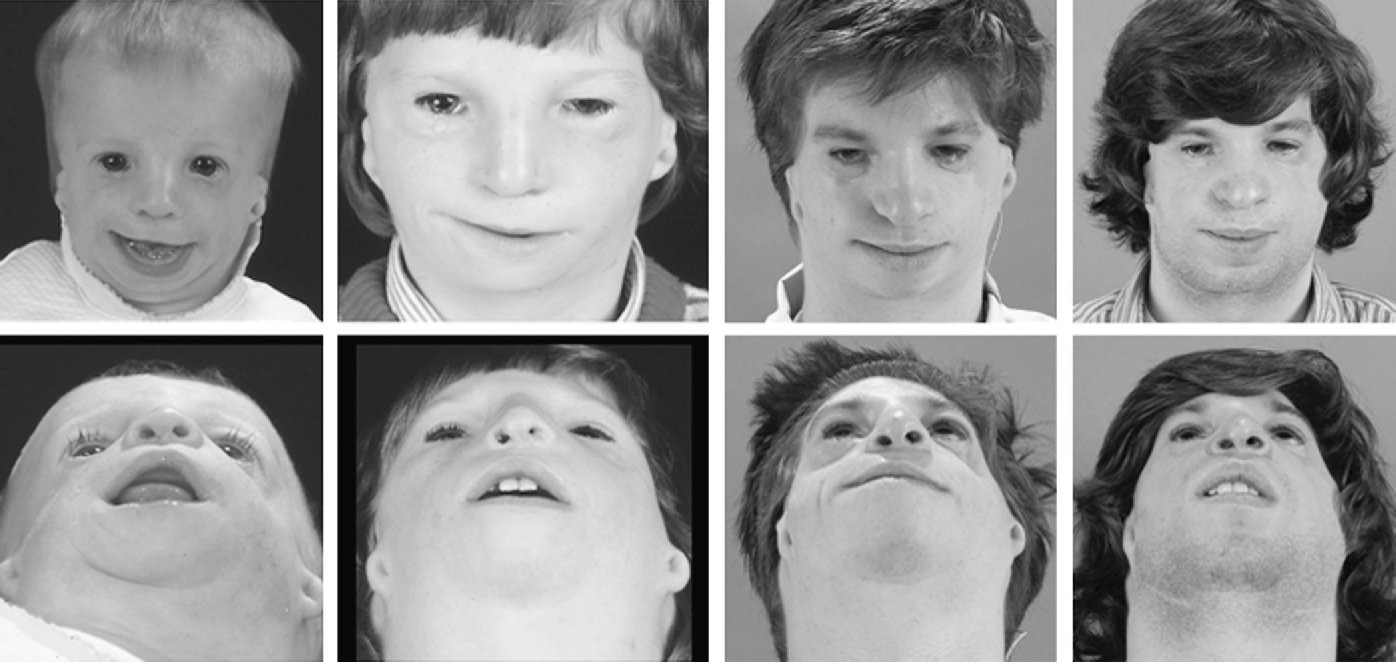
Стадии
Учитывая всю проблематичность мутаций, проявления данного недуга могут быть выражены очень ярко, или же быть незначительными.
Классифицируют такие стадии патологического процесса:
- На первом этапе видоизменения практически незаметны. Такие пациенты счастливчики, ведь они попали именно в ту категорию, которая позволяет им вести полноценный образ жизни.
- На второй стадии признаки налицо, как в прямом, так и в переносном смысле. Также могут возникнуть и сложности с функцией дыхания и функционированием других органов.
- Одной из наиболее сложных является третья стадия. Она сопровождается почти полной деформацией лица. При этом никакая терапия и пластика не будут эффективны.
Лечение синдрома Франческетти
К сожалению, необходимо констатировать тот факт, что в настоящее время медицина не готова предложить терапевтические методы лечения данной проблемы, их попросту не существует. Вся терапия направлена исключительно на паллиативную помощь. Человек, который отличается от всех остальных, сильно желает быть кому-то нужным. Поэтому в этом случае ценнее простой человеческой заботы трудно что-либо найти.

Если наблюдаются тяжёлые стадии заболевания, то следует делать операцию. Если у человека при этом есть проблемы со слухом, врачи рекомендуют использовать слуховой аппарат. Конечно, от моральной поддержки зависит очень многое. Сможет ли пациент поверить в себя? Сможет ли быть сильнее всех проблем? Довольно трудные вопросы. Это говорит о том, что психологическая помощь не может быть лишней никогда. В любой ситуации при возможности необходимо поддержать добрым словом. Вам это ничего не стоит, а больному это может открыть второе дыхание.
Осложнения
Одним из самых серьезных последствий синдрома Тричера принято считать недоразвитие ротового аппарата. Значительная деформация зубов, челюстей и отсутствие слюнных желез приводят к отсутствию у больных способности принимать самостоятельно пищу. Кроме того, врожденная аномалия может провоцировать появление заболеваний дыхательной системы по причине больших размеров языка и зарастания носовых проходов.
- Офломелид — инструкция по применению и состав, форма выпуска, показания и цена
- Токсоплазмоз головного мозга — причины возникновения, симптомы и лечение
- Токсоплазмоз у детей — симптомы и лечение, профилактика заражения
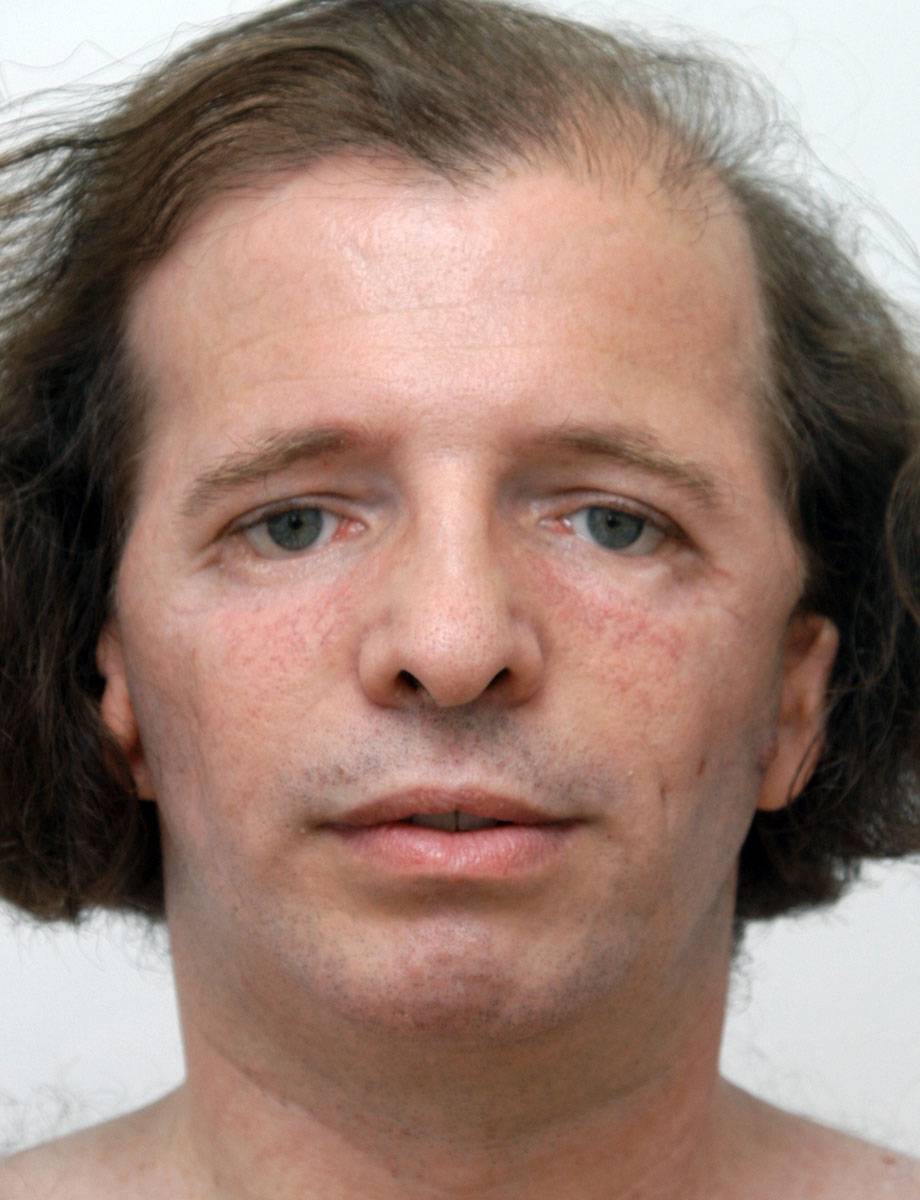
Что такое синдром Тричера Коллинза?
Внешние мутации при синдроме Тричера Колллинза могут иметь разную степень выраженности: от незначительных отклонений развития лобной, лицевой, носовой костей до тяжёлых патологий. При этом заболевание затрагивает только черепно-лицевой скелет, без каких-либо нарушений умственного и физического развития больного.
Информация
Впервые подобные патологии в самом начале ХХ века описал Эдвард Тричер Коллинз, ранее прославившийся своими трудами в области анатомии и патологии глаз. Впоследствии, синдром получил имя этого английского хирурга и офтальмолога. Позже более подробные труды проводил офтальмолог из Швеции Адольф Франческетти, введший понятие «мандибуло-фасциального дизостоза». В европейских медицинских кругах заболевание носит двойное название – синдром Франческетти-Коллинза – от имён обоих исследователей.
Основные
нарушения развития скелета лица, вызванные мутацией гена TCOF1, происходят на раннем сроке беременности (3-4 неделя).
Диагностика
Стоит отметить, что выявление данного недуга происходит во время беременности. У сформировавшегося плода врачи диагностируют наличие синдрома Франческетти. Рекомендуется проводить исследование лицевой части с помощью биопсии. Однако этот способ не является безопасным, и поэтому врачи часто используют для этих целей УЗИ. Также необходимо у всех членов семьи взять анализы крови. После этого беременной женщине следует пройти фетоскопию. Заключительным этапом диагностики является взятие крови из плацентарных сосудов.
Причины синдрома Франческетти кроются в мутации генов, поэтому в зависимости от выраженности болезни врачи принимают решение о целесообразности дальнейших исследований. Если заболевание характеризуется экспрессивностью, то здесь сомнений быть не может. Однако в случае обнаружения некоторых отдельных симптомов специалисты обязаны продолжить наблюдение. Список мероприятий для выявления синдрома составляется врачом индивидуально, и в большинстве случаев это приносит свои плоды
Данное заболевание является довольно тяжёлым, и поэтому очень важно диагностировать его заранее, чтобы оставался шанс на исправление положения вещей
Признаки синдрома
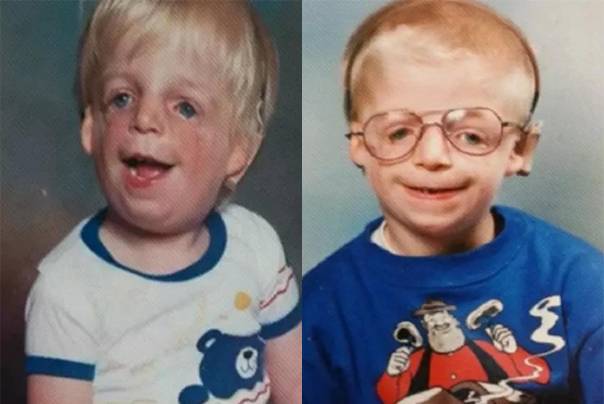
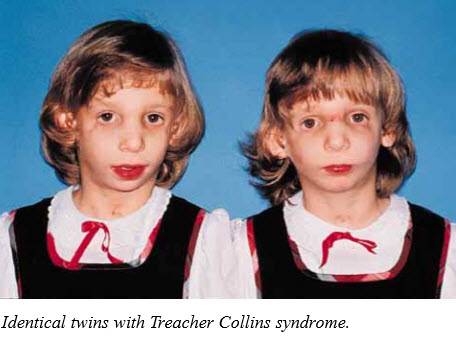
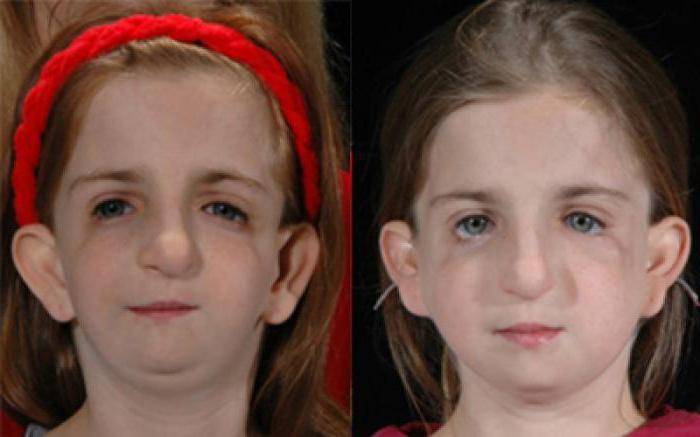
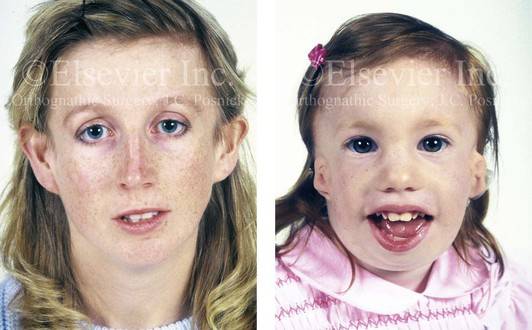
Так как данная болезнь характеризуется тем, что поражает ещё не родившийся плод, то первые симптомы можно обнаружить сразу после появления на свет. Более того, практически все признаки недуга проявляются сразу. Даже несведущий человек в медицине при мимолётном взгляде на больного может выявить определённые симптомы. Главным из них является нарушение формы глаз. Видно сразу человека с синдромом Франческетти — фото, представленные в статье, это наглядно демонстрируют.

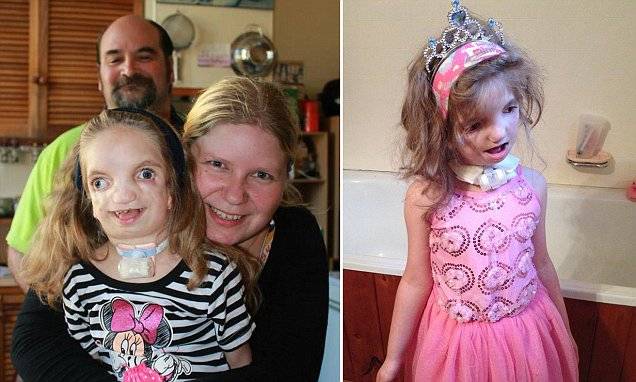

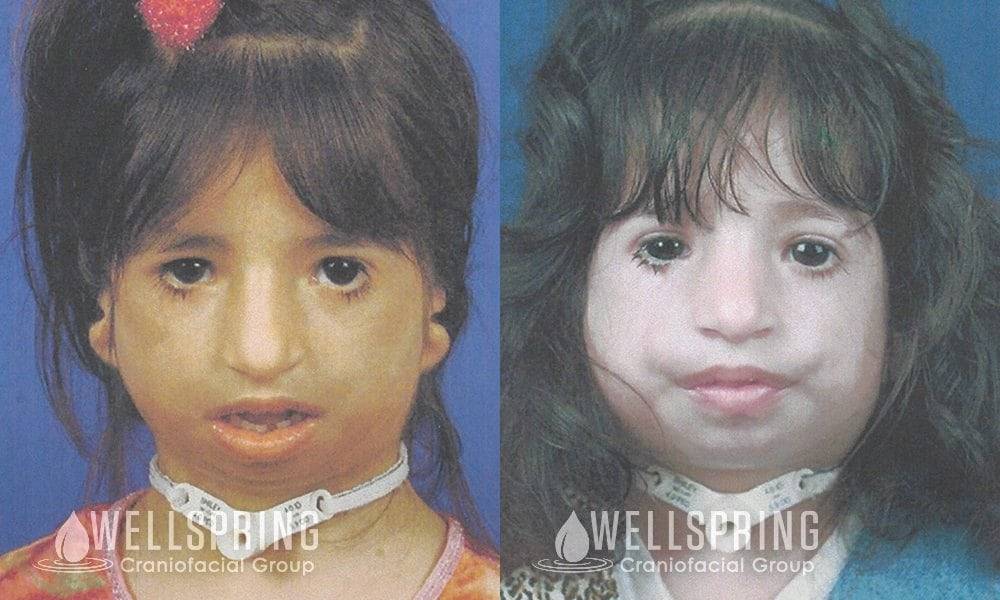
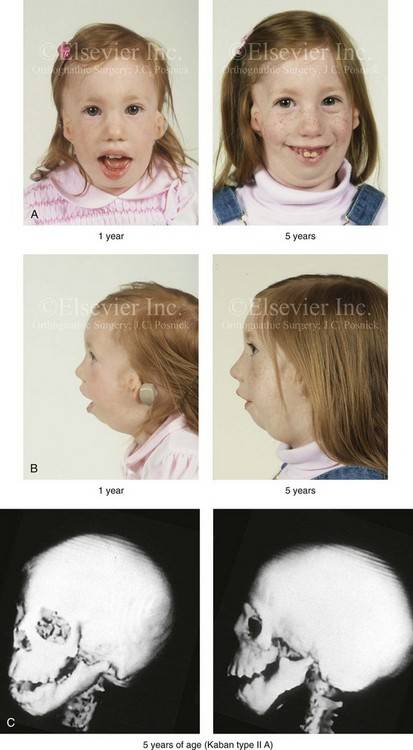
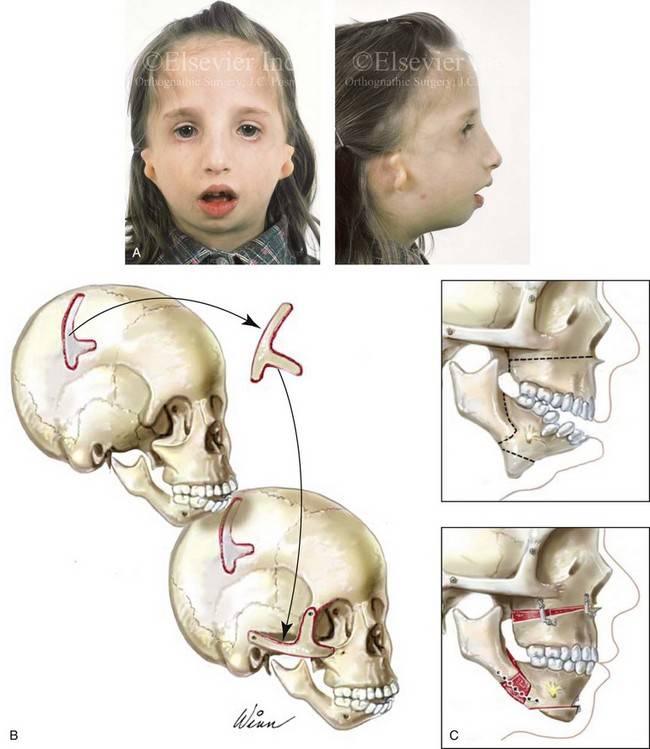
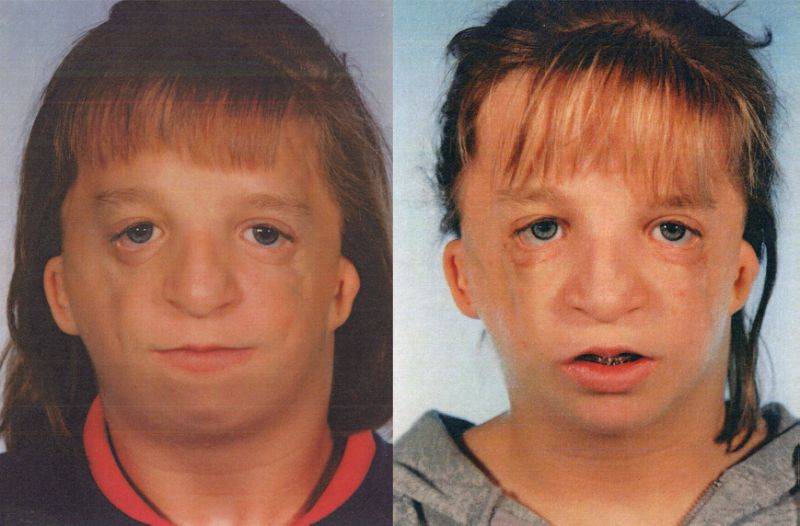




Кроме вышеуказанного признака, есть ещё несколько, которые характеризуют данное заболевание. Стоит отметить, что все симптомы довольно ярко выражены, и их сложно не заметить. Итак, среди них наиболее распространёнными являются:
- сбой в строении скул и нижней челюсти;
- проблемы со слухом;
- нарушение в структуре подбородка;
- отсутствие ушных раковин;
- неправильный прикус.
Почему появляется заболевание
Несмотря на то, что данная патология наследуется от больных родителей, в 60% случаях происходит первичное развитие новых мутаций, которые вызваны следующими негативными факторами:
- злоупотребление алкогольными напитками;
- воздействие радиации;
- цитомегаловирусная инфекция и токсоплазмоз;
- контакт с токсическими химическими веществами;
- прием некоторых лекарственных средств (психотропные, противоэпилептические препараты, а также с содержанием ретиноевой кислоты).
Их влияние на организм женщины до беременности или во время нее может вызвать возникновение дефекта в генах.
Уровни развития недуга
В начале статьи было указано, что существует несколько стадий болезни, о которых пришло время поговорить подробнее. Синдром Франческетти (Коллинза) имеет три уровня развития. Первый и наиболее безопасный характеризуется небольшой гипоплазией костей лицевой части. Однако даже в этом случае человек меняется внешне, и самые внимательные могут заподозрить наличие заболевания.
Когда наступает вторая стадия, то здесь уже добавляются проблемы со слухом, неправильная форма глазных щелей, деформация нижней челюсти. В этом случае недуг становится более явным, и человек с синдромом внешне значительно отличается от обычного индивида.
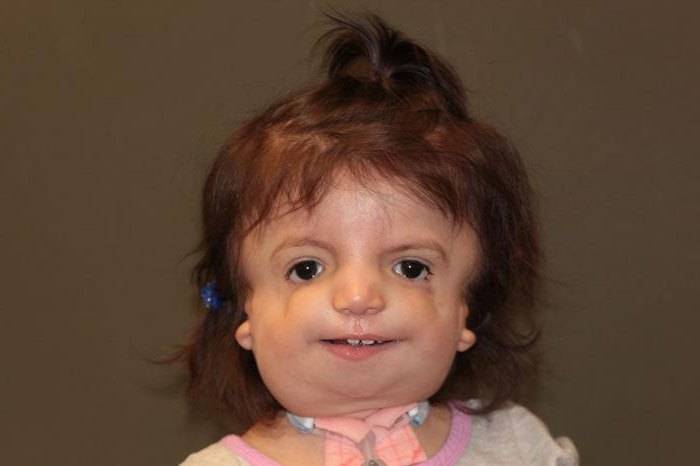
Что касается последней, наиболее тяжёлой стадии, то тут имеет место полное отсутствие лица. На самом деле это очень страшно, ведь пациент просто автоматически исключается из общества, и с очень низкой долей вероятности что-то изменится. Кроме того, эти люди с годами страдают сильнее, ведь патология только усугубляется. Могут появиться признаки более тяжёлых болезней. Синдром Франческетти – сам по себе довольно опасный недуг, а с годами могут приобретаться заболевания и похуже.