Первичные дефициты клеточного иммунитета
К первичным дефицитам клеточного иммунитета относятся следующие заболевания:
- Синдром Ди Джорджи
- Синдром Дункана
- Недостаточность пуриннуклеозидфосфорилазы
- Оротацидурия
- Биотин-зависимые ферментопатии.
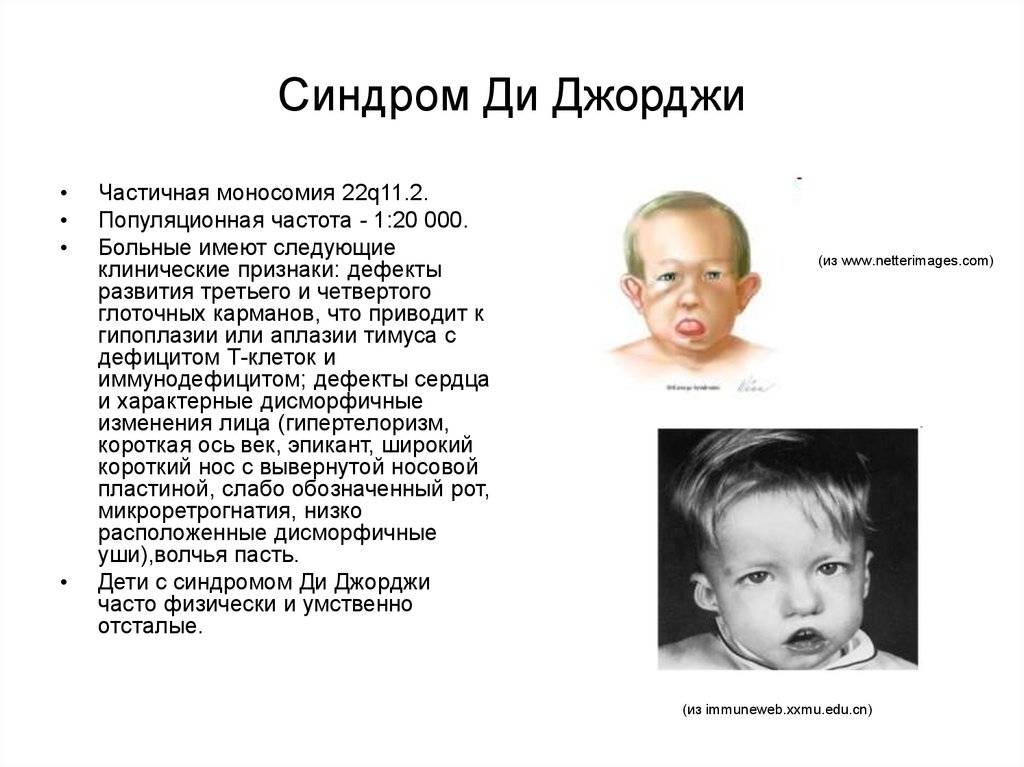
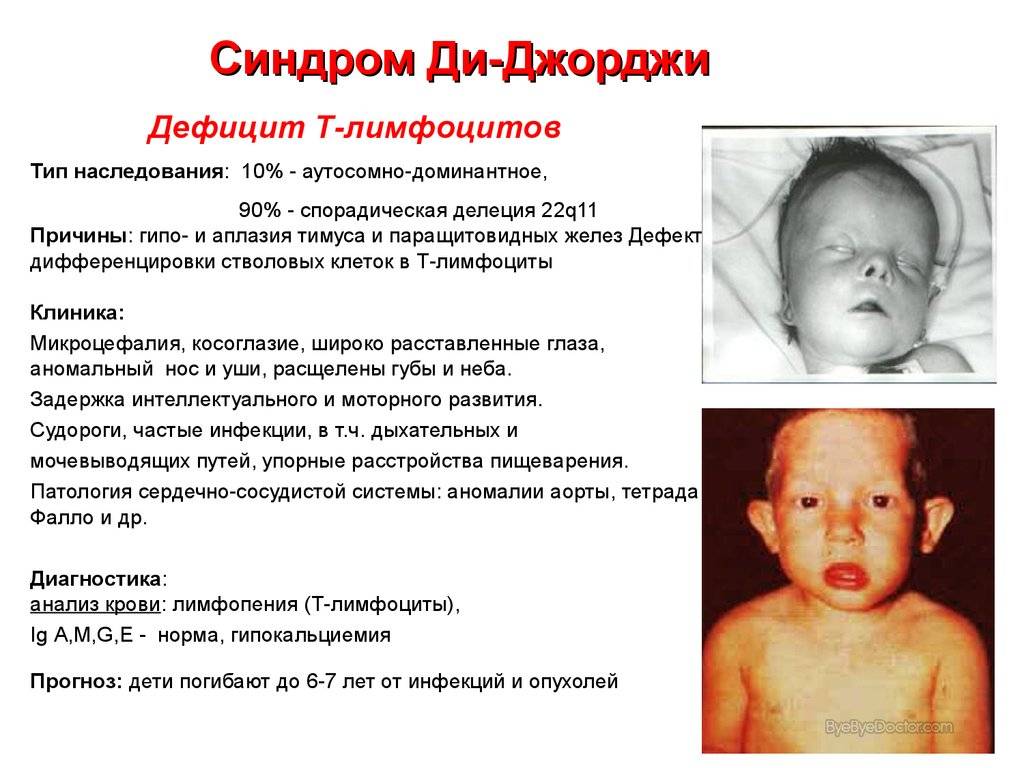
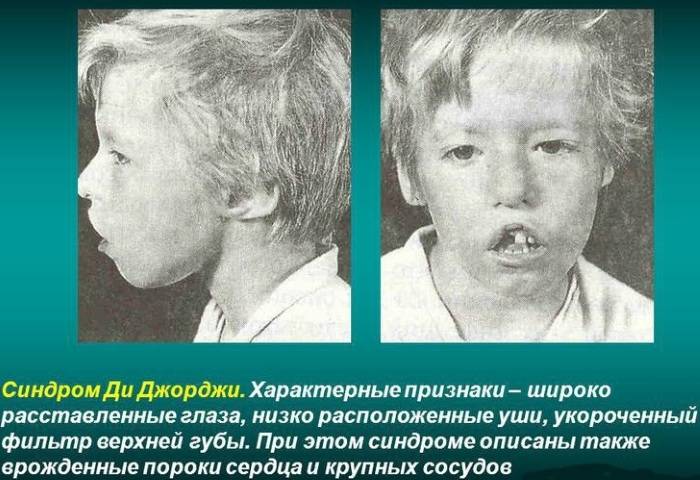
Синдром Ди Джорджи
В основе синдрома Ди Джо́рджи (Di George) лежит гипоплазия тимуса. Синдром описан в г. Считается, что это заболевание не является наследственным, оно возникает в результате приобретённого нарушения органогенеза в области III—V жаберных дуг (глоточных карманов) на 6—8 неделе беременности. Поэтому, кроме порока тимуса, отмечаются дефекты околощитовидных желёз, сердца и крупных сосудов, а также орофациальные пороки (микростомия, микрогнатия, гипертелоризм, низкое расположение ушных раковин).
Результатом гипоплазии паращитовидных желёз является дефицит парат-гормона и персистирующая гипокальциемия, вследствие чего развивается судорожный синдром, который может проявиться уже в первые часы жизни (неонатальная тетания). Причиной смерти детей в более старшем возрасте служат осложнения, связанные с пороками развития сердца.
Нарушения, затрагивающие Т-лимфоциты, могут быть как очень глубокими, так и едва заметными. В любом случае функция Т-клеток с возрастом восстанавливается и к 5 годам, если ребёнок остаётся жив, не удаётся обнаружить их недостаточности. Антиген-независимый этап созревания Т-клеток при этом происходит вне тимуса — в многослойных плоских эпителиях, прежде всего в эпидермисе. Одним из эффективных способов лечения синдрома Ди Джорджи является трансплантация эмбриональной ткани тимуса.
Синдром Дункана
Синдром Ду́нкана (Х-сцепленный лимфопролиферативный синдром) — иммунодефицит, характеризующийся повышенной чувствительностью к вирусу Эпштейна—Барр. Ген повышенной чувствительности к вирусу локализован в Х-хромосоме, тип наследования заболевания рецессивный, поэтому болеют мальчики. У больных, перенёсших инфекционный мононуклеоз, развиваются длительное лихорадочное состояние, лимфаденопатия (увеличение лимфатических узлов), лимфоцитоз периферической крови, гепато- и спленомегалия. Позднее формируется В-клеточная лимфома, чаще в терминальных отделах тонкой кишки, от которой больные и погибают. Летальные исходы обусловлены также деструктивным гепатитом, вызываемым вирусом Эпштейна—Барр.
Недостаточность пурин-нуклеозид-фосфорилазы
Недостаточность пурин-нуклеозид-фосфорилазы (ПНФ) наследуется по аутосомно-рецессивному типу. Дети страдают гипопластической анемией и крайне сниженной функцией Т-клеток.
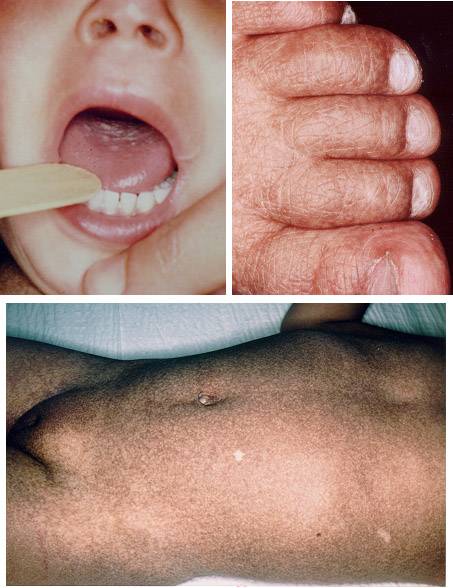
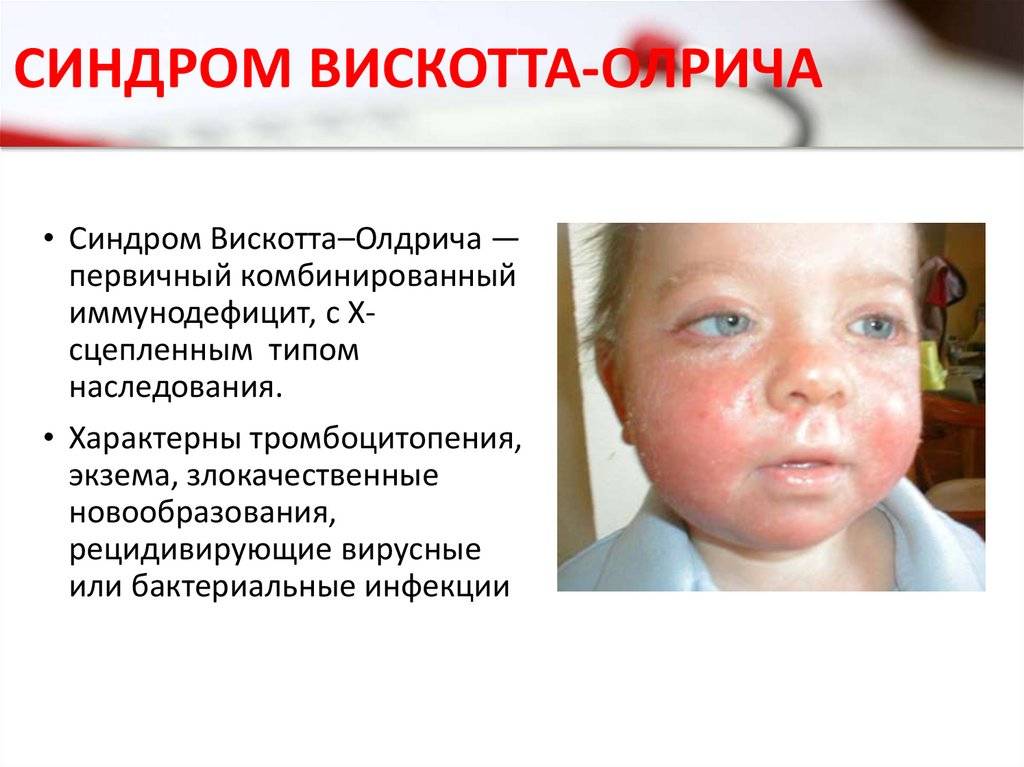
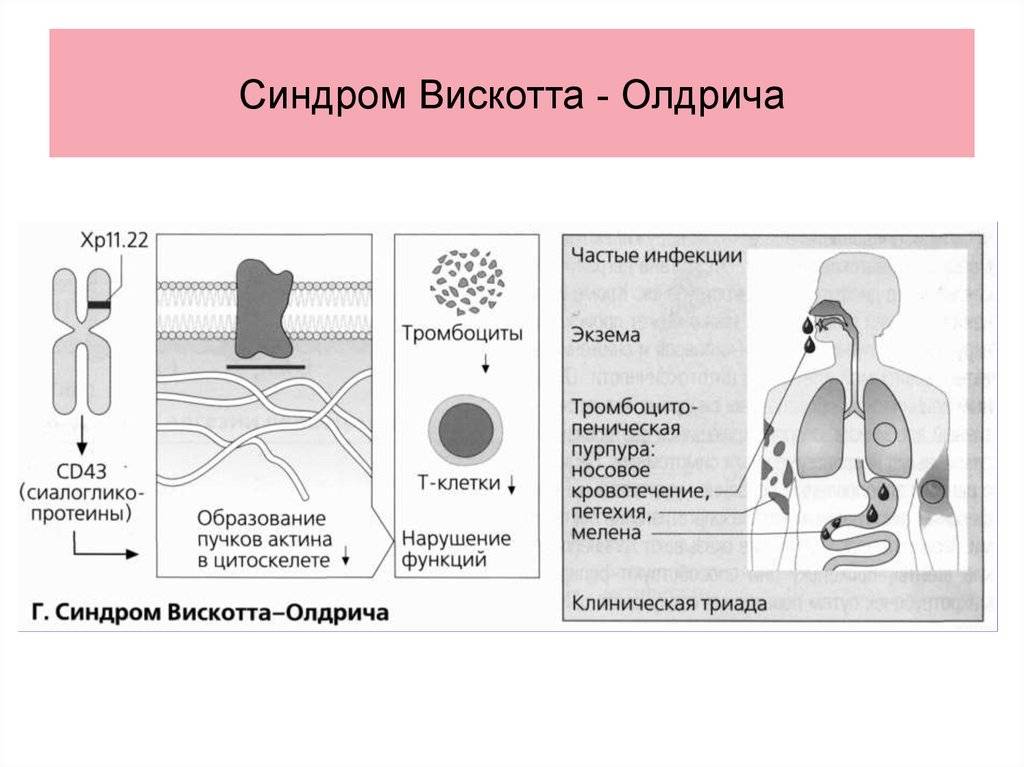
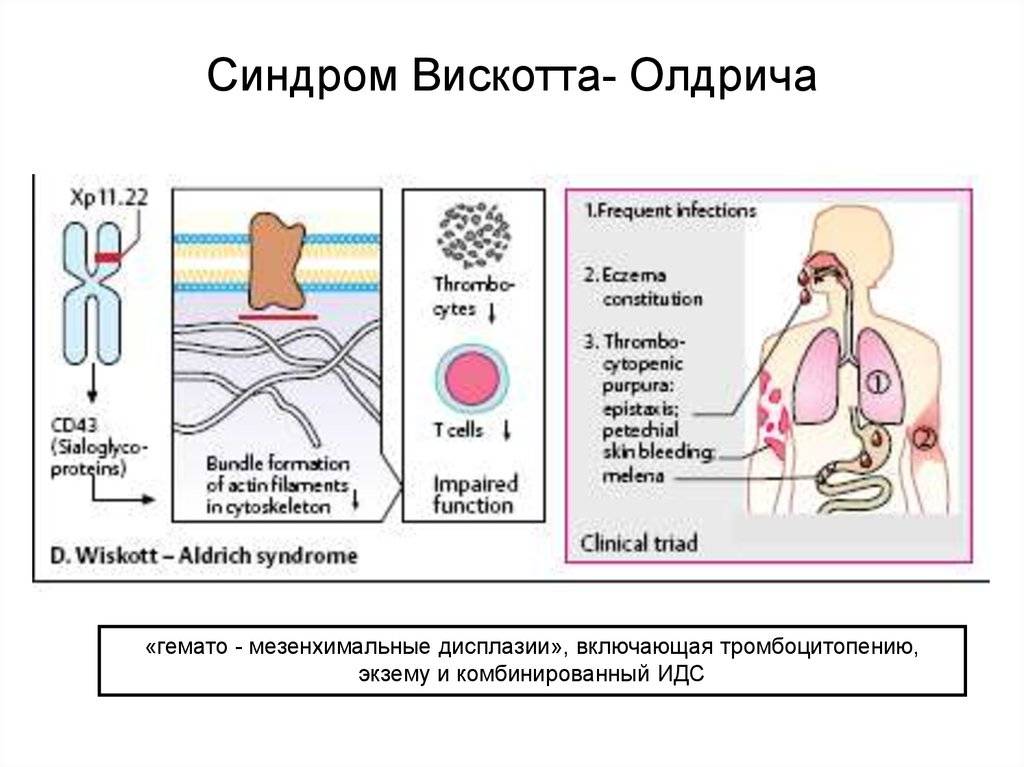


Оротацидурия
Оротацидури́я — наследственное заболевание синтеза пиримидинов, которое проявляется повышенной экскрецией оротовой кислоты (оротата) с мочой, недостаточностью Т-лимфоцитов, мегалобластной анемией и задержкой умственного и физического развития. При этом заболевании снижена активность ферментов оротидил-пирофосфорилазы и оротидил-декарбоксилазы, которые преобразуют оротовую кислоту в нуклеотид-оротидин-монофосфат, необходимый для синтеза нуклеиновых кислот.
Биотин-зависимые ферментопатии
Биотин-зависимые ферментопатии также сопровождаются развитием клеточного иммунодефицита (наследственные дефекты биотинидазы и биотин-зависимых энзимов пируват-карбоксилазы и пропионат-карбоксилазы, участвующих в метаболизме аминокислот с разветвлённой цепью — валина, лейцина, изолейцина). Заболевание проявляется уже в периоде новорождённости эпизодами кетоацидоза, неврологической симптоматикой, алопецией, кожными сыпями и непереносимостью белка (рвота, мальдигестия, дегидратация). В моче содержится большое количество органических кислот. Дети отстают в физическом развитии. Из инфекционных процессов наиболее часто развиваются кандидоз и кератоконъюнктивиты. Биотин даёт хороший терапевтический эффект.
Транзиторная гипогаммаглобулинемия у детей
Патогенез.
Характеризуется гипогаммаглобулинемией
вследствие нарушения образования IgG у
детей раннего возраста и диагностируется
после исчезновения материнских
трансплацентарных IgG. Транзиторная
гипогаммаглобулинемия сохраняется у
детей с 6 мес до 2-3 лет. Уровень IgG при
этом снижен в два раза по сравнению с
возрастной нормой при нормальных или
сниженных показателях IgA и IgM.
Клиника.У некоторых младенцев симптомы не
проявляются. Они нормально отвечают на
антигены вакцин и спустя несколько лет
«перерастают» гипогаммаглобулинемию.
У других детей выявляют рецидивирующие
бактериальные инфекционные заболевания
начиная с первого месяца жизни. Основные
клинические проявления — бактериальные
инфекции верхних дыхательных путей. У
некоторых детей выявляют рецидивирующую
диарею, тяжелые формы ветряной оспы,
длительно сохраняющийся оральный
кандидоз. У большинства детей развиваются
аллергические заболевания. Лимфатические
узлы и миндалины у таких детей
гипоплазированы.
Диагностика:Уровень сывороточных IgA и IgG обычно
снижен, а IgM чаще в норме или повышен.
Содержание В-лимфоцитов в норме,
нейтропения и реже тромбоцитопения. У
большинства детей симптомы транзиторной
гипогаммаглобулинемии исчезают к 2-3
годам. Повторно определять уровни
иммуноглобулинов необходимо с интервалом
6-12 мес, пока не восстановятся нормальные
показатели.
Лечение
симптоматическое, направленное на
купирование инфекций. В тяжелых случаях
показана заместительная терапия
препаратами иммуноглобулинов.





Идс с недостаточностью в-клеточного звена иммунитета и антител
Развиваются вследствие неспособности
В-клеток продуцировать иммуноглобулины,
характеризуются
– появлением симптомов в возрасте 7–9
месяцев, после исчезновения материнских
АТ
– повторными инфекционными заболеваниями,
вызванными инкапсулированными бактериями
– хроническими очагами инфекции
(синуситы, бронхиты, пневмонии), гнойными
лимфаденитами, абсцессами
– менингитами, септицемией, остеомиелитом,
возникающими в результате гематогенного
распространения патогена
– редкой заболеваемостью грибковыми
и вирусными инфекциями (за исключение
энтеровируса)
– выживаемостью до зрелого возраста
при адекватном лечении
– повышенная частота аллергических и
аутоиммунных заболеваний
– гипоплазия периферических лимфатических
узлов и назофаренгеальной лимфоидной
ткани (характерно для синдрома Брутона)
– лимфоидная гиперплазия (гепатоспленомегалия
при ОВИД)
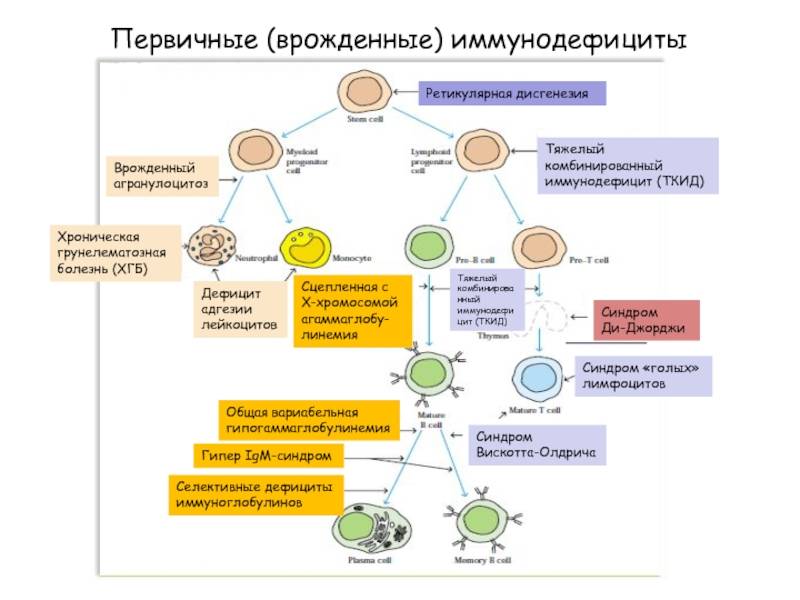
Первичные иммунодефициты
Определение и классификация
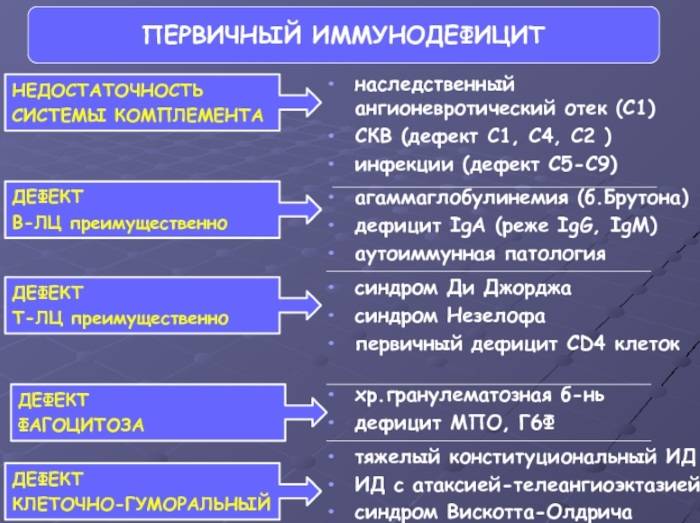
Первичные иммунодефициты — это врожденные (генетические или эмбриопатии) дефекты иммунной системы. В зависимости от уровня нарушений и локализации дефекта они бывают:
- гуморальные или антительные — с преимущественным поражением системы В-лимфоцитов
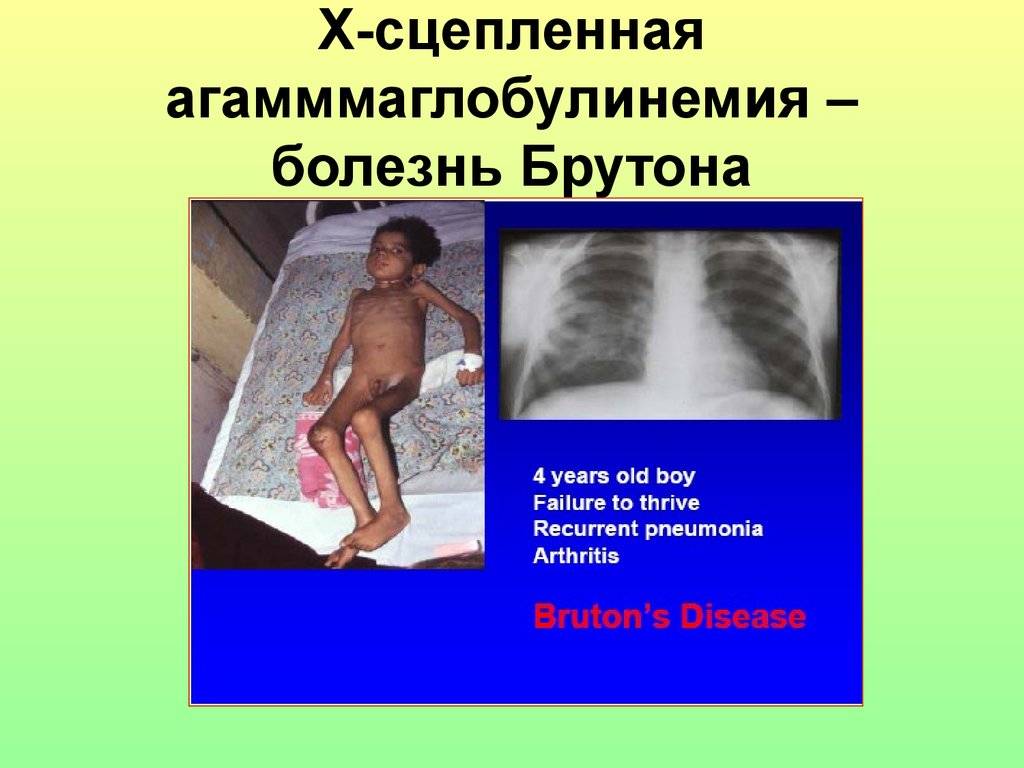
- Х-сцепленная агаммаглобулинемия (болезнь Брутона)
- Гипер-IgM синдром
- Х-сцепленная
- аутосомно-рецессивная
- делеция генов тяжелых цепей иммуноглобулинов
- дефицит k-цепей
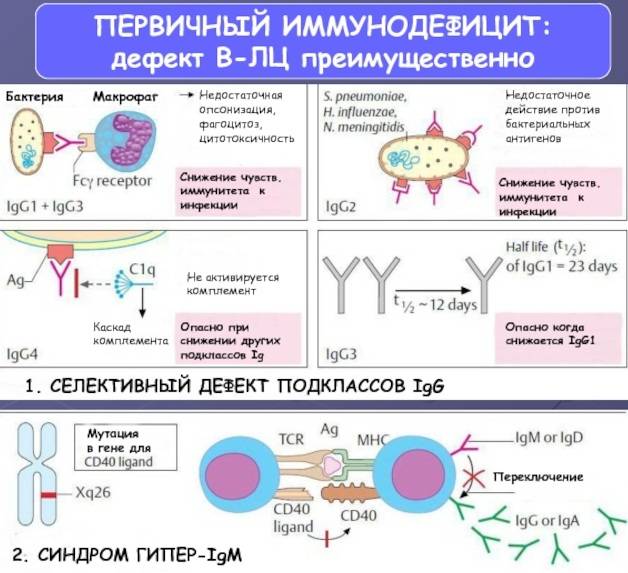
- селективный дефицит субклассов IgG с или без дефицита IgA
- дефицит антител с нормальным уровнем иммуноглобулинов
- общая вариабельная иммунная недостаточность
- дефицит IgA
- клеточные
- синдром Ди Джоржи
- первичный дефицит CD4 клеток
- дефицит CD7 Т-клеток
- дефицит ИЛ-2
- множественная недостаточность цитокинов
- дефект передачи сигнала
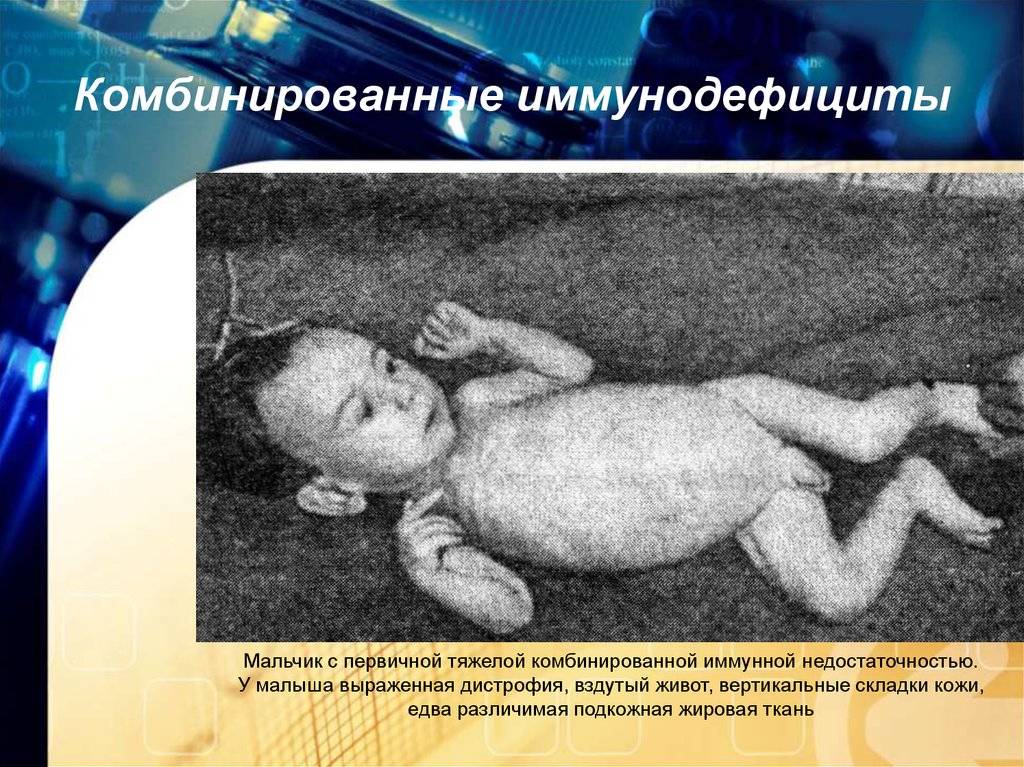
- комбинированные:
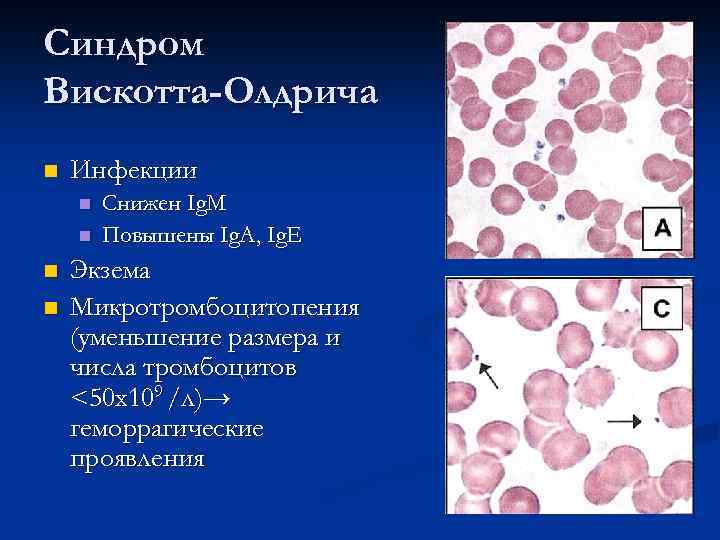
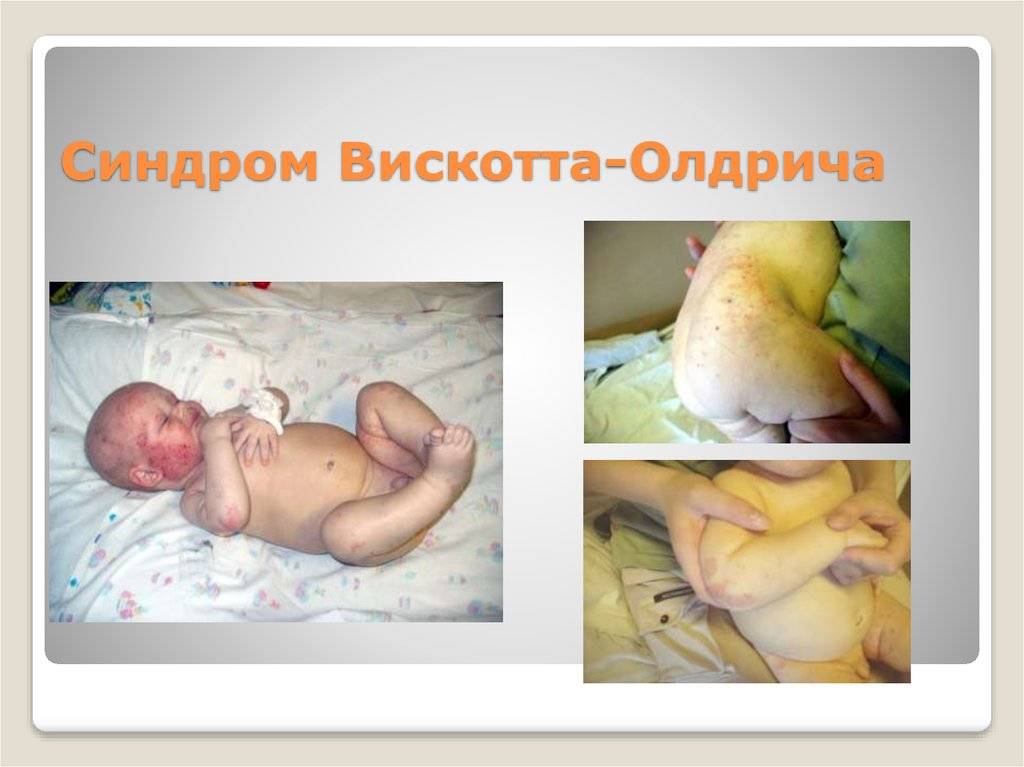
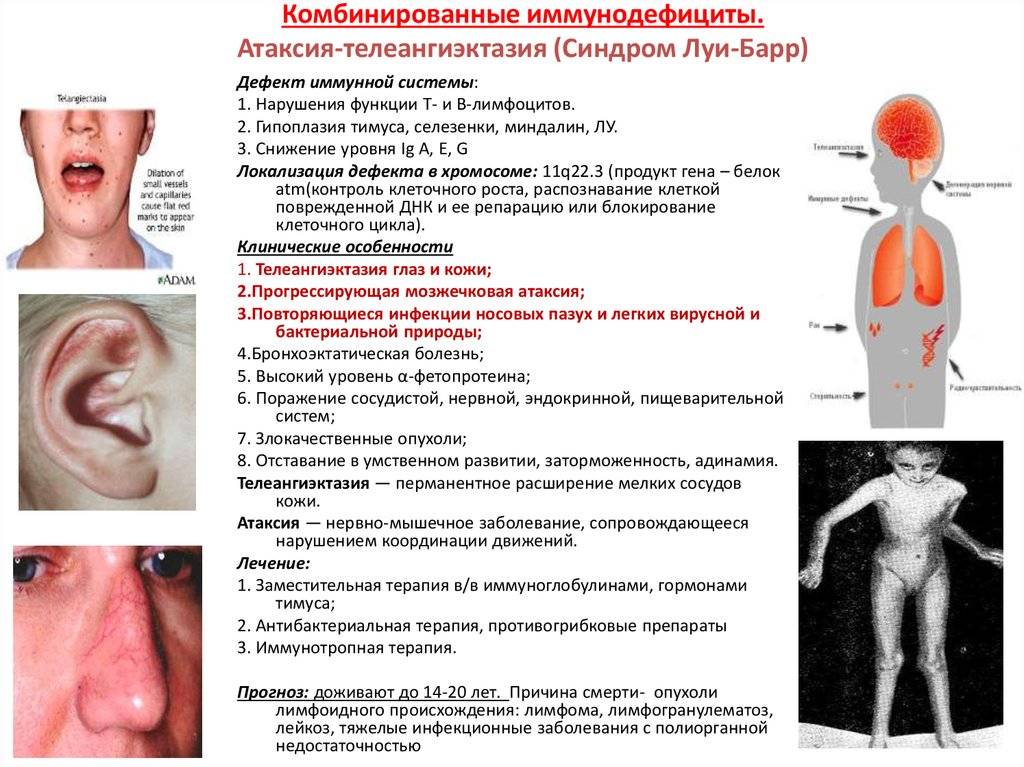
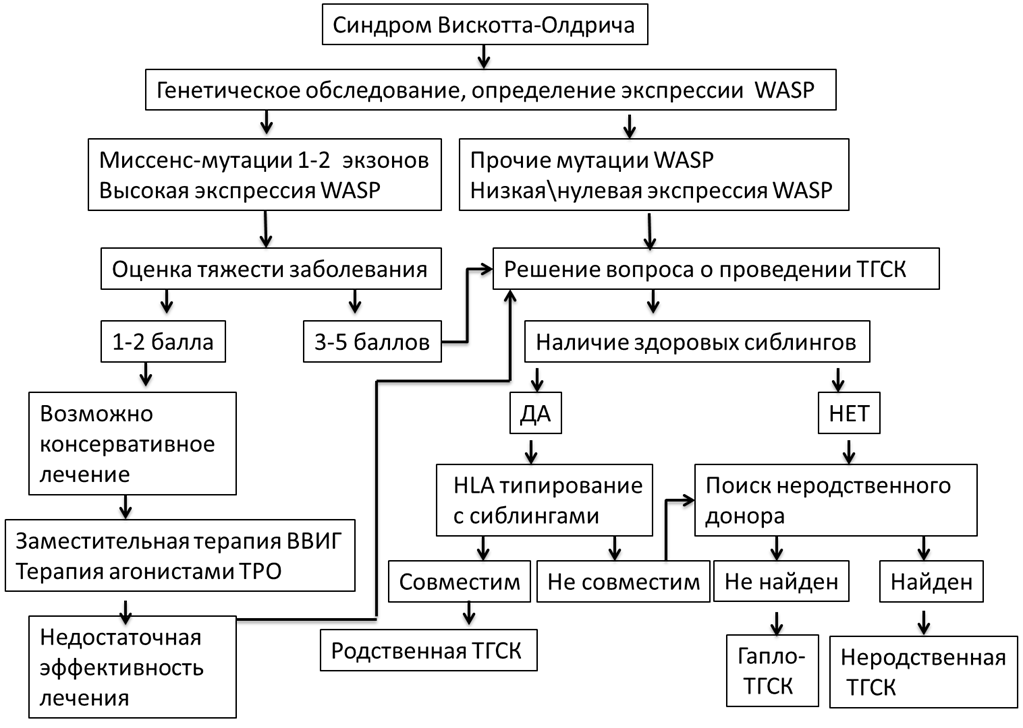
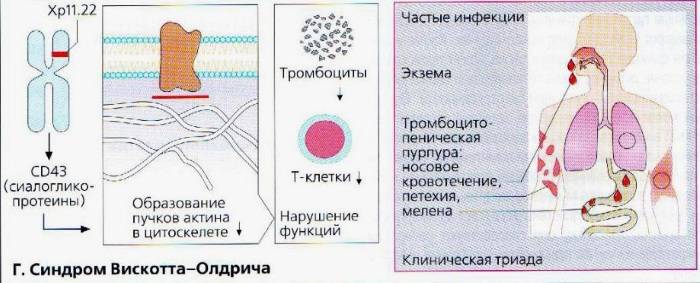
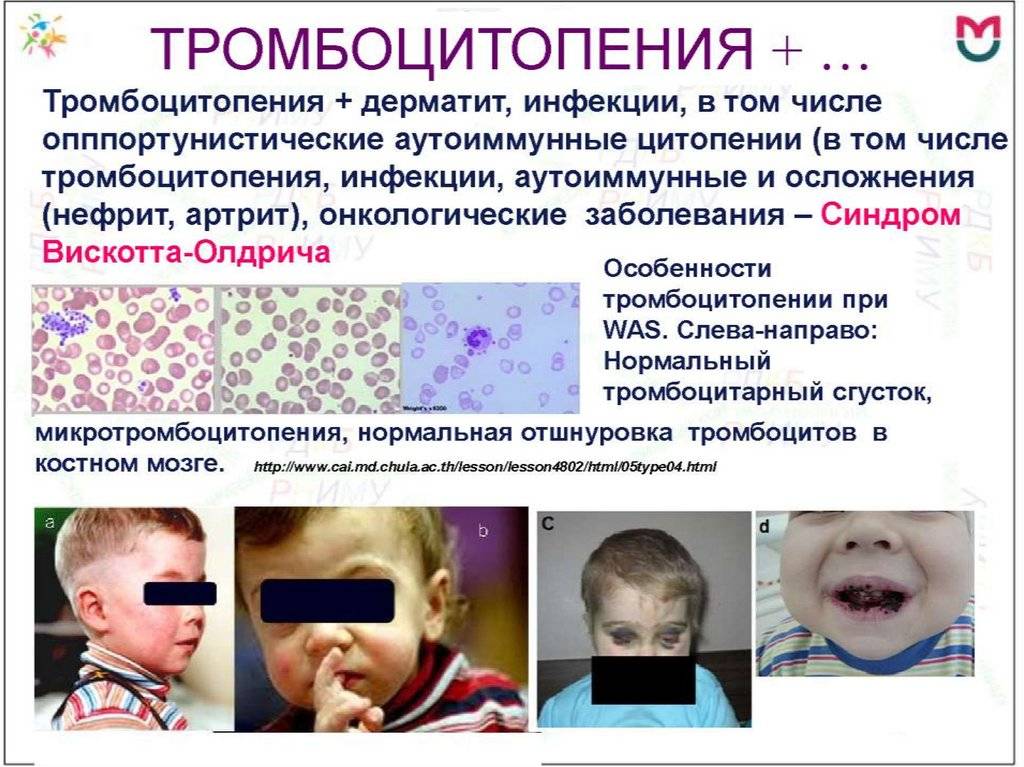
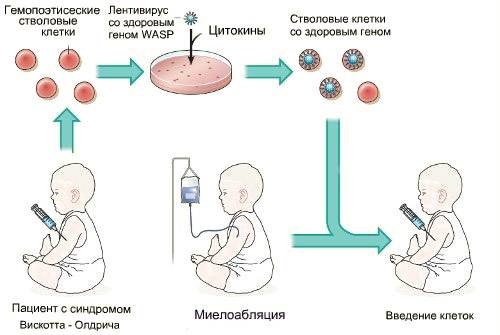
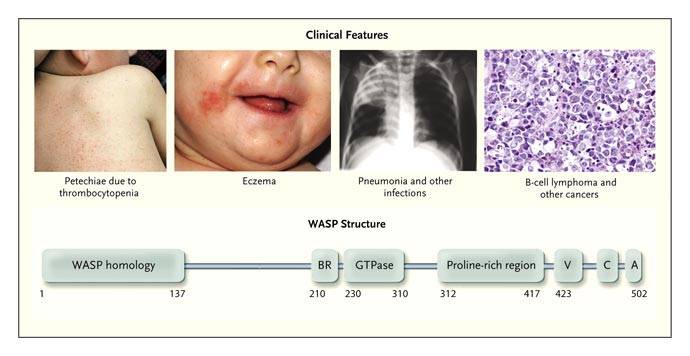
- синдром Вискотта-Олдрича
- атаксия-телеангиоэктазия (синдром Луи-Бар)
- тяжелая комбинированная иммунная недостаточность
- Х-сцепленная с полом
- аутосомно-рециссивная
- дефицит аденозиндезаминазы
- дефицит пуриннуклеозидфосфорилазы
- дефицит молекул II класса МНС (синдром лысых лимфоцитов)
- ретикулярная дизгенезия
- дефицит CD3γ или CD3ε
- дефицит СD8 лимфоцитов
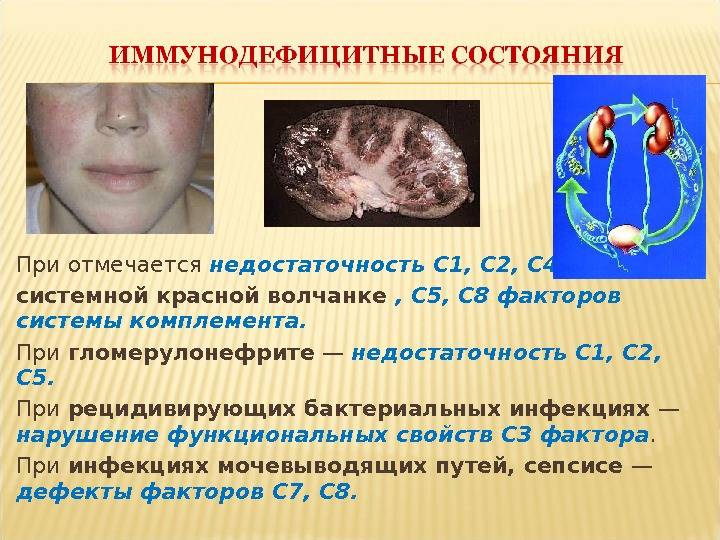
- недостаточность системы комплемента
- дефекты фагоцитоза
- наследственные нейтропении
- инфантильный летальный агранулоцитоз (болезнь Костмана)
- циклическая нейтропения
- семейная доброкачественная нейтропения
- дефекты фагоцитарной функции
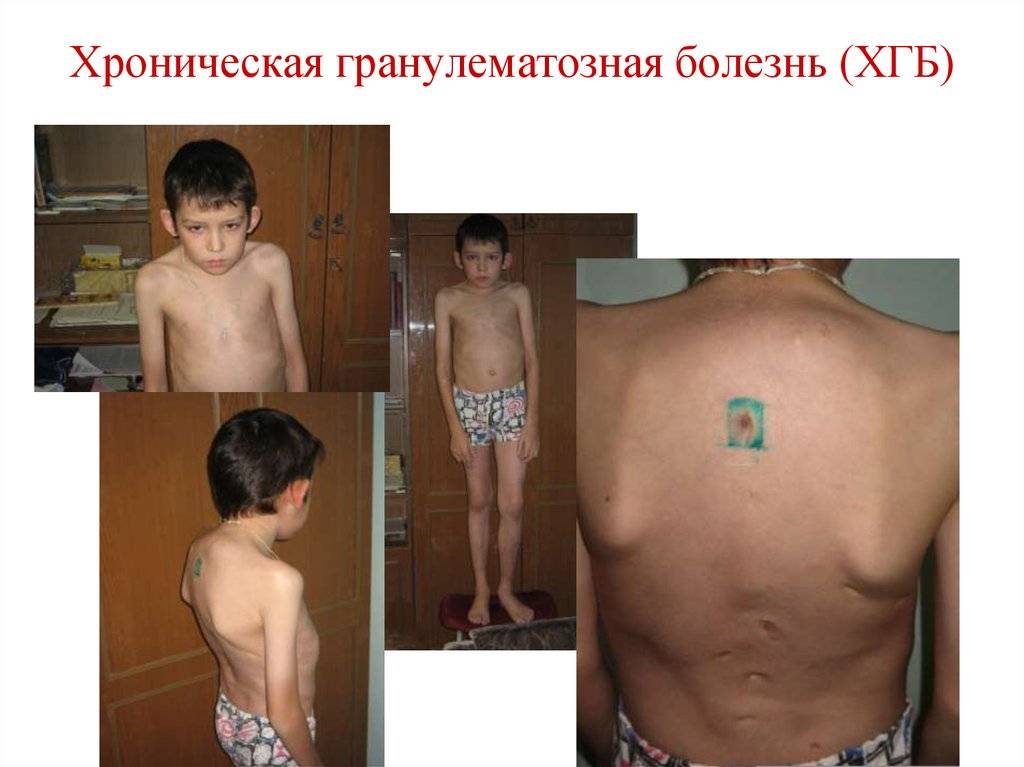
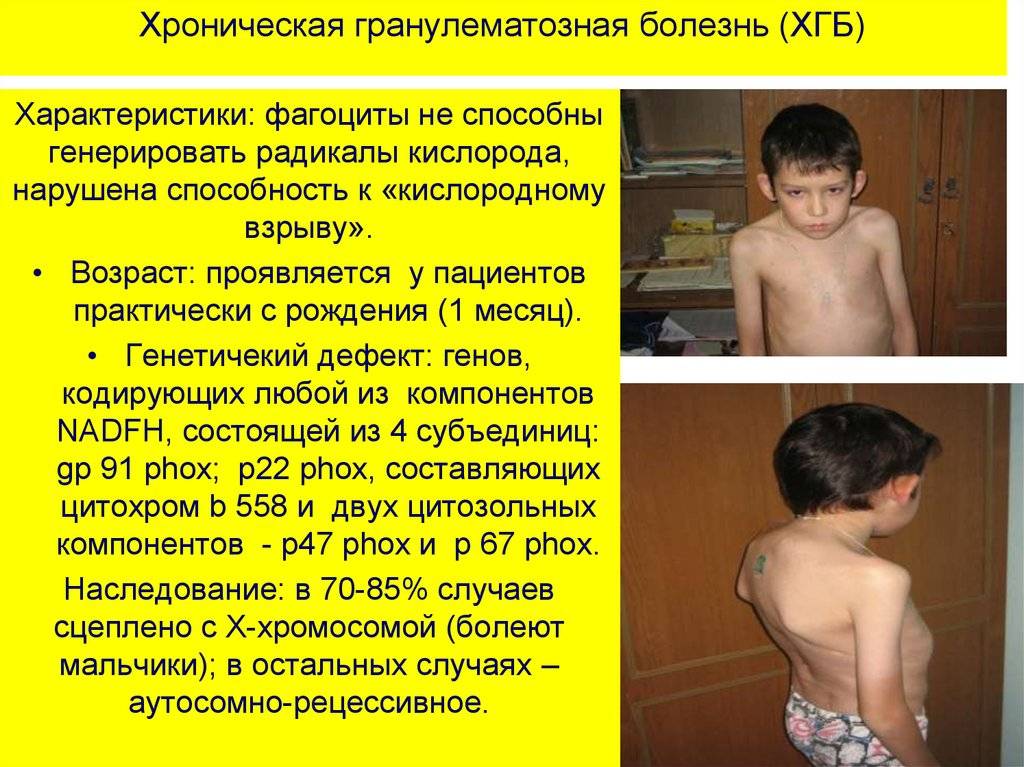
- хроническая гранулематозная болезнь
- Х-сцепленная
- аутосомно-рециссивная
- дефицит адгезии лимфоцитов I типа
- дефицит адгезии лейкоцитов 2 типа
- дефицит глюкозо-6-дегидроегназы нейтрофилов
- дефицит миелопероксидазы
- дефицит вторичных гранул
- синдром Швахмана
- наследственные нейтропении
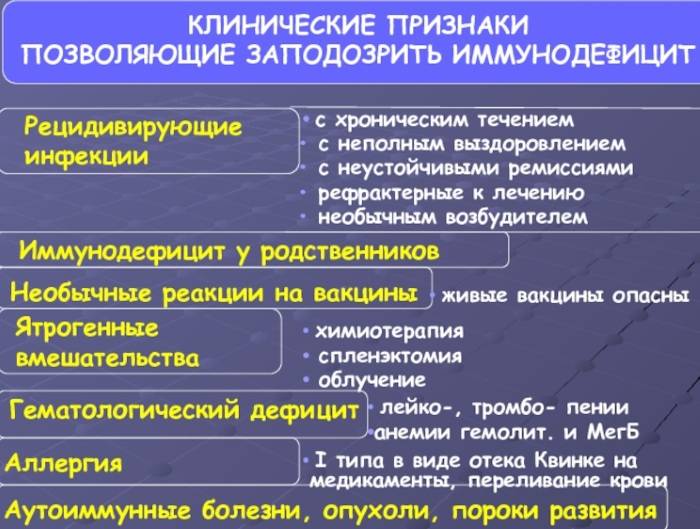
Клиническая картина ИДС
Клиника имеет ряд общих черт:
- 1. Рецидивирующие и хронические инфекции верхних дыхательных путей, придаточных пазух, кожи, слизистых оболочек, желудочно-кишечного тракта, часто вызываемые оппортунистическими бактериями, простейшими, грибами, имеющие тенденцию к генерализации, септицемии и торпидные к обычной терапии.
- 2. Гематологические дефициты: лейкоцитопении, тромбоцитопении, анемии (гемолитические и мегалобластические).
- 3. Аутоиммунные расстройства: СКВ-подобный синдром, артриты, системная склеродермия, хронический активный гепатит, тиреоидит.
- 4. Нередко ИДС сочетается с аллергическими реакциями 1 типа в виде экземы, отека Квинке, аллергическими реакциями на введение лекарственных препаратов, иммуноглобулина, крови.
- 5.Опухоли и лимфопролиферативные заболевания при ИДС встречаются в 1000 раз чаще, чем без ИДС.
- 6. У больных с ИДС часто отмечаются расстройства пищеварения, диарейный синдром и синдром мальабсорбции.
- 7. Больные с ИДС отличаются необычными реакциями на вакцинацию, а применение у них живых вакцин опасно развитием сепсиса.
- 8. Первичные ИДС часто сочетаются с пороками развития, прежде всего, с гипоплазией клеточных элементов хряща и волос. Кардиоваскулярные пороки описаны, главным образом, при синдроме Ди-Джоржи.
Лечение первичных ИДС
Этиотропная терапия заключается в коррекции генетического дефекта методами генной инженерии. Но такой подход является экспериментальным.
Основные усилия при установленном первичном ИДС направлены на:


- профилактику инфекций
- заместительную коррекцию дефектного звена иммунной системы в виде трансплантации костного мозга, замещения иммуноглобулинов, переливания нейтрофилов.
- заместительную терапию ферментами
- терапию цитокинами
- витаминотерапию
- лечение сопутствующих инфекций
- генная терапия
- иммуномодулирующя терапия
В 2018 году российский препарат на основе высокоочищенных прошел . В ходе испытаний была подтверждена безопасность применения лекарственного средства. Планировалось, что после регистрации и завершения дополнительных исследований, препарат возможно будет применять в качестве заместительной и иммуномодулирующей терапии у пациентов со сниженным или отсутствующим уровнем синтеза антител. Средство направлено на обеспечение нормализации уровня иммуноглоублина до оптимальных значений и повышение сопротивляемости организма к патогенам.
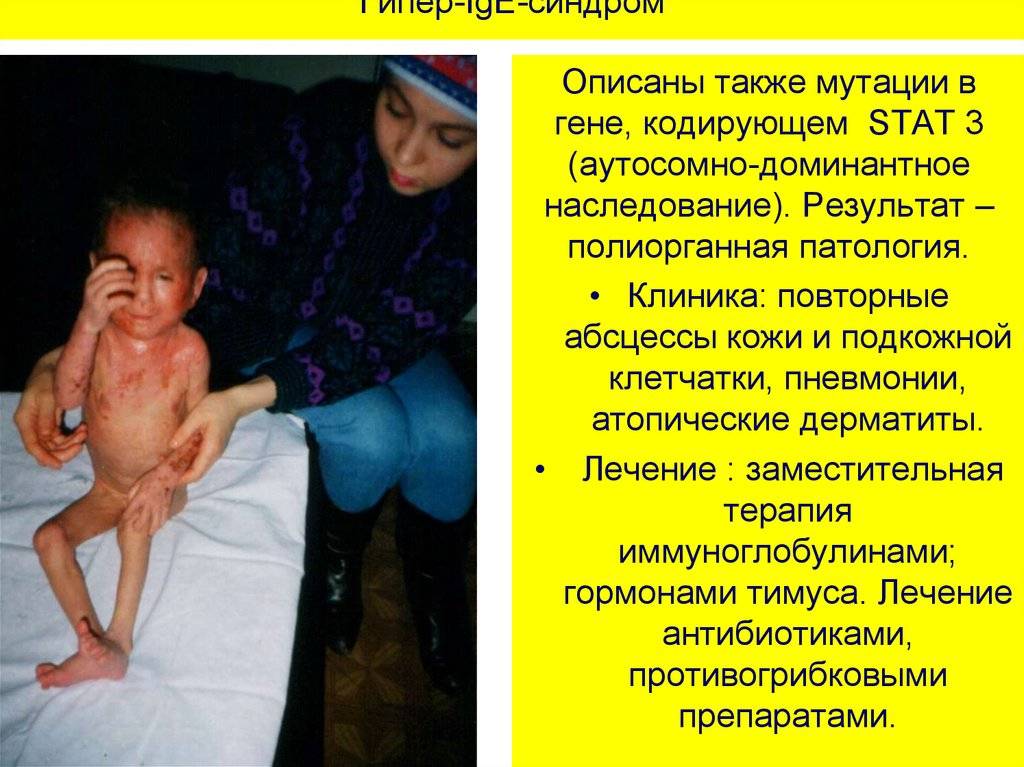
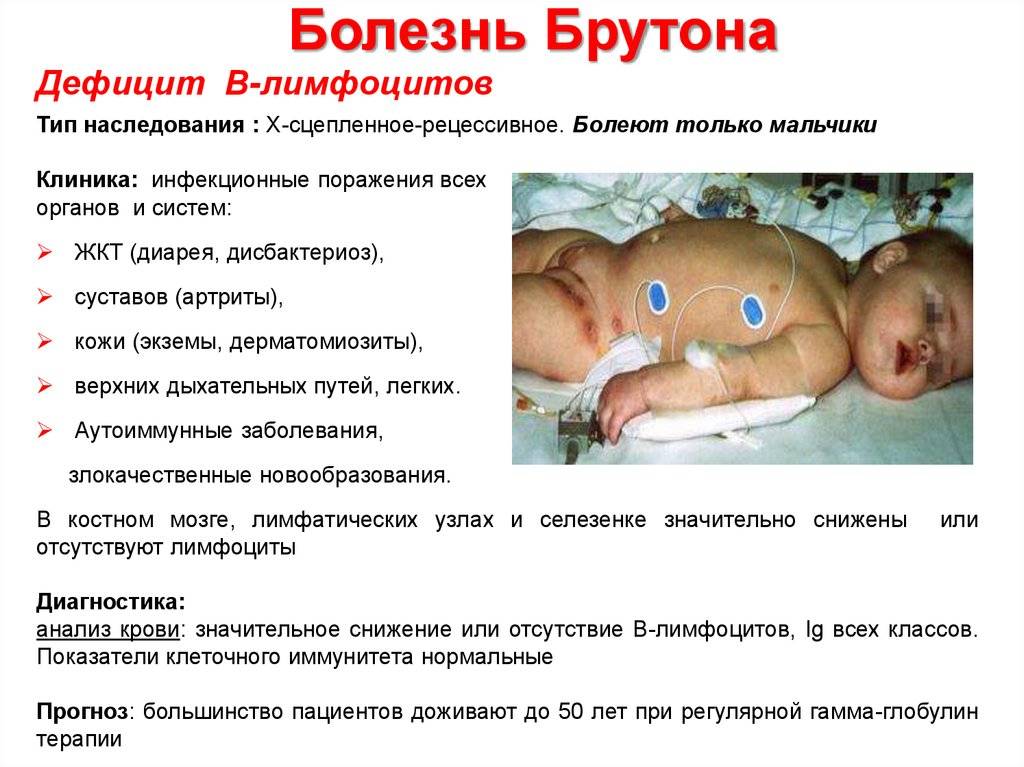
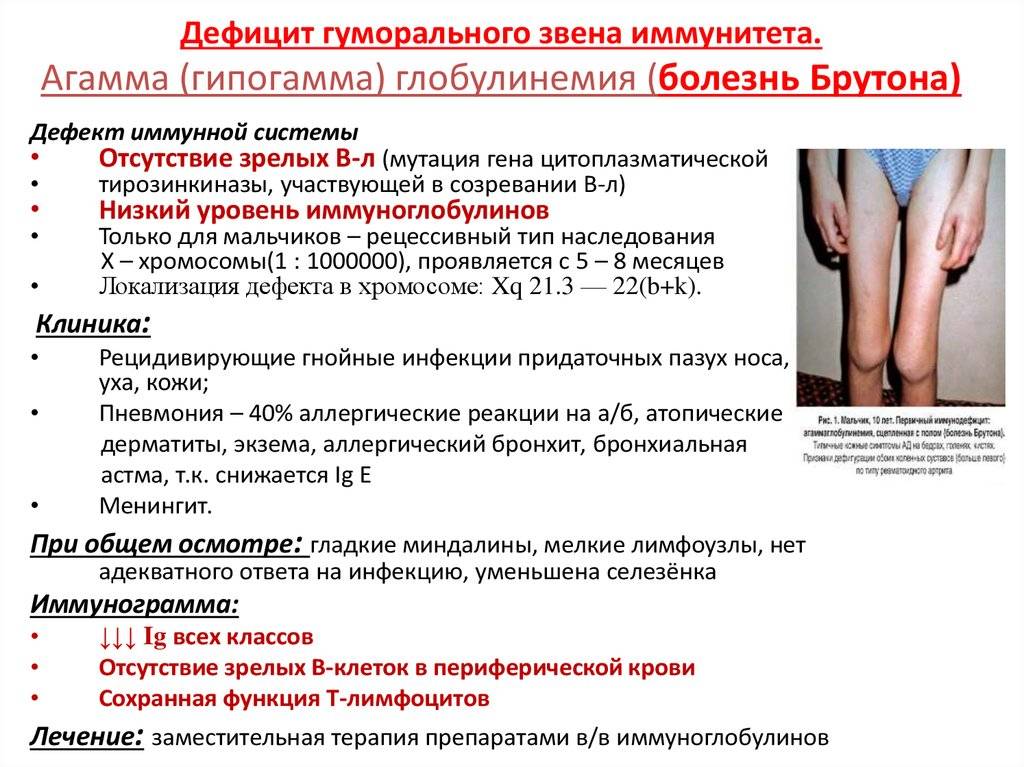
Сидром Брутона
(сцепленная с полом агаммаглобулинемия
у детей)
Тип наследованияи частота«классической» формы — рецессивный,
сцепленный с Х-хромосомой. Заболевание
наблюдается только у мальчиков. Частотой
1 : 50 000.
Генетический дефект и патогенез.
Дефектный ген (Xq22)
кодирует цитоплазматическую тирозинкиназу
В-клеток (Btk). В результате
дефекта нарушено превращение пре-В-клеток
в зрелые В-клетки.
Клинические проявления. Заболеваниепроявляется с 7-8 мес. жизни (так как
начинает разрушатьсяIgGполучаемый от матери). При этом заболевании
снижена резистентность организма к
стафилококку, стрептококку, пневмококку,
а также к некоторым грамотрицательным
микроорганизмам (кишечной палочке,
сальмонеллам, протею, клебсиелле), часто
наблюдаются грибковые заболевания, а
также паразитарные (пневмоцистные)
пневмонии. Детичастоболеют
рецидивирующими пневмониями, отитом,
синуситом, конъюктивитом, пневмонией,
пиодермией, которые нередко приводят
к развитию сепсиса. Низкая сопротивляемость
к бактериальным инфекциям не сочетается
со снижением резистентности к вирусам.
Некоторые вирусные заболевания (краснуха,
корь, вирусный гепатит) у них протекают
даже легче, чем у детей с сохраненной
иммунологической реактивностью.
Диагностика. Общее содержаниеIgGв крови ребенка составляет менее 2 г/л.
Особенно резко снижено содержание IgG и
IgА. При антигенной стимуляции (например,
при вакцинации АКДС) отсутствует
нарастание титров соответствующих
антител. Показатели, характеризующие
клеточный иммунитет, не отличаются от
таковых в норме. Отмечено уменьшение
или отсутствие лимфоцитов и плазмоцитов
в костном мозге, последние не содержатся
в лимфатических узлах и селезенке.
Прогноз по сравнению с другими ИДС
относительно благоприятный. Пациенты
обычно достигают возраста 20 лет.
Продолжительность жизни во многом
зависит от тяжело протекающих вирусных
инфекций (полиомиелита, энтеровирусная),
а также от лимфоретикулярных опухолей
(около 6 % случаев).