Диагностические мероприятия
Визит к офтальмологу затягивать не рекомендуется, поскольку очень велик риск потери зрения
Если у пациента возникают нарушения зрительной функции, важно точно определить их причину. Медик проведет опрос пациента, во время которого выясняет, насколько давно возникли проблемы со зрением и имеются ли подобные патологические симптомы у близких родственников
В случае, когда подобные проблемы обнаруживаются у членов семьи, диагностический процесс протекает, как правило, достаточно легко. Если же типичной симптоматики заболевания у родственников пациента не наблюдается, диагностические мероприятия существенно затрудняются.
В первую очередь при этом необходимо исключить заболевания нервной системы. Наибольшую сложность во время диагностики представляют большие группы миопатических синдромов, спровоцированные эндогенными и экзогенными и патологиями. К примеру, миопатия зрительных органов часто возникает при первичном амилоидозе и миоглобинурии. Для постановки правильного диагноза пациенту требуется пройти такие обследования:
- анализ ферментов крови;
- электромиография;
- биопсия мышечной ткани.
Диагностика миопии, как следствия данного патологического процесса, обычно не составляет особого труда и предусматривает проведение следующих мероприятий:
- проверка зрения с помощью таблиц Сивцева;
- периметрия (изучение полей зрения);
- офтальмоскопия глазного дна;
- онометрия (измерение внутриглазного давления);
- ультразвуковое исследование;
- офтальмометрия (определение оптической силы роговицы);
- скиаскопия (изучение движения тени в зоне зрачка);
- тонография (изучение продукции и процессов оттока внутриглазной жидкости).
Лечение заболевания в зависимости от его формы
Воспалительные миопатии лечатся курсом глюкокортикостероидов. Доза Преднизолона на сутки равна 80-100 мг. Если эффект от лечения становится заметен (в мышцах появляется сила), дозу постепенно снижают до поддерживающей – 15 мг/сут.
В осложненных случаях воспалительная миопатия требует проведения метилпреднизолоновой пульс-терапии. Лечение гормональными препаратами имеет множество побочных эффектов, поэтому глюкокортикостероиды некоторым больным противопоказаны.
Если в течение 3-х месяцев лечения глюкокортикостероидами положительная динамика не наблюдается, значит, миопатия приобрела стероидную резистентность. В этом случае пациенту назначают цитостатики:
- Циклоспорин;
- Азатиоприн;
- Циклофосфамид;
- Метотрексат.
Лечение миопатии Шарко-Мари является симптоматическим. Больному назначают витамины, группы В, АТФ, антихолинэстеразные медикаменты, сеансы физиотерапии, переливания крови, ЛФК, массаж. Терапия проводится повторными курсами.
Если у пациента свисающие стопы, он должен носить ортопедическую обувь. В тяжелых случаях показана тенотомия. При этом заболевании профессиональная деятельность больного не должна быть связана с большими нагрузками.
Лечение миопатии Дюшена – обычно малоэффективно, что обусловлено быстрым прогрессированием болезни и большой ее тяжестью.
Пациенту назначают поддерживающую терапию, которая смягчает симптоматику и немного замедляет процесс развития недуга. Это препараты для улучшения метаболизма:
- аминокислоты;
- витамины В, Е;
- Оксазил;
- Калия оротат;
- анаболические гормоны;
- кальциевые препараты;
- Галантамин;
- Прозерин.
Лечение должно проходить курсами и в условиях стационара. Применение лекарств группы глюкокортикоидов на несколько лет продлевает больным жизнь.
Лечение миопатии Дюшена
В настоящее время вылечить эту болезнь невозможно. Однако существует ряд способов, которые могут облегчить состояние пациента на разных стадиях развития миопатии и, возможно, несколько замедлить прогрессирование заболевание.
Ниже описаны способы лечения для определенных возрастных групп, но нередко при лечении одного пациента приходится сочетать сразу несколько методов терапии.
Пациенты дошкольного возраста
Обычно в этом возрасте детям с миопатией Дюшена еще не нужно лечение. Родителям могут предложить:
Подробную информацию о миопатии Дюшена. Врач проведет с родителями беседу и подробно расскажет о том, как эта болезнь будет влиять на состояние ребенка, и какова ожидаемая продолжительность жизни пациента (большинство людей с таким диагнозом доживают лишь до 20-25 лет). При желании родители могут обратиться к другим специалистам (например, к психологу), а также в группы поддержки;
Рекомендации относительно допустимых физических нагрузок для ребенка;
Генетическую консультацию для членов семьи. Многие родители желают узнать, являются ли они носителями гена Дюшена
Это особенно важно для тех, кто в будущем планирует снова родить ребенка.
Уже в дошкольном возрасте ребенка начинают регулярно обследовать, чтобы, когда это будет необходимо, можно было вовремя начать лечение.
Пациенты в возрасте 5-8 лет
Детям такого возраста может потребоваться поддержка для мышц ног. Например, им могут рекомендовать надевать на ночь шины для лодыжки или более длинные шины, для голени.
При помощи кортикостероидов можно замедлить развитие миопатии и в течение некоторого времени сохранять мышцы достаточно сильными. Пациенты постоянно или курсами принимают такие препараты, как преднизолон или дефлазакорт. Поскольку кортикостероиды могут вызывать серьезные побочные эффекты, ребенок обязательно должен наблюдаться у врача.

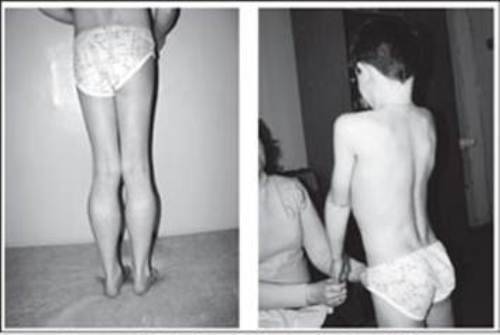
Пациенты от 8 лет до позднего переходного возраста
Через какое-то время после достижения ребенком возраста восьми лет его мышцы начинают заметно слабеть. Ходить со временем становится все труднее пока, наконец, ребенку не придется начать передвигаться на инвалидном кресле. Возраст, в каком это происходит, варьируется от пациента к пациенту. Часто это случается между 9 и 11 годами, но дети, которые достаточно рано начали принимать кортикостероиды, иногда могут продолжать ходить несколько дольше.
Вскоре после того, как ребенку для передвижения становится необходимым инвалидное кресло, у него начинают развиваться и другие осложнения, поэтому ему могут потребоваться более частые обследования. Все осложнения необходимо начинать лечить как можно раньше.
Кроме этого, родителям нужно позаботиться о практической стороне жизни ребенка – на первых порах помогать ему передвигаться в кресле, а также по возможности приспособить свой дом под его нужды.
Пациенты от позднего переходного возраста до 20+ лет
В этом возрасте мышечная слабость вызывает все больше проблем, и пациенту все чаще требуется помощь других людей. Увеличивается вероятность развития тяжелых осложнений, таких как легочные инфекции.
Прогноз
Как говорилось выше, миопатия Дюшена – тяжелое заболевание, которое значительно сокращает жизнь человека. Со временем мышечная слабость вызывает все более серьезные проблемы с дыхательной системой и работой сердца. В прошлом большинство пациентов с миопатией Дюшена доживали лишь до 20-23 лет. Сегодня все больше людей с этим диагнозом доживают до 27 лет, а иногда и до более старшего возраста. Отметим, что продолжительность жизни зависит от многих факторов, таких как сопутствующие заболевания, доступность качественной медицинской помощи, и так далее. Со временем ожидаемая продолжительность жизни при миопатии Дюшена может еще больше увеличиться.
Наиболее распространенной причиной смерти больных являются осложнения, связанные с респираторной системой, например, тяжелые инфекции дыхательных путей.
Классификация миопатий
На сегодняшний день единой классификации миопатий не существует, эти заболевания подразделяют по нескольким принципам. Первой работой в этом направлении была так называемая клиническая классификация нервно-мышечных заболеваний, считающихся в те времена болезнями исключительно мышечных тканей. Согласно этой системе, медики выделяли такие типы миопатий, как конечностно-поясная, лице-плече-лопаточная, гумеро-тибиальная и другие.
Данное деление можно было считать условным, так как оно не отличалось четкостью характеристик, как и сегодня. Более точной классификации миопатий и нервно-мышечных заболеваний в медицине нет, поэтому до сих пор применяется существующая система.
Известна также патогенетическая классификация, появление которой связано с возникновением новых знаний о миопатии. К примеру, стало известно, что миодистрофии могут проявляться по причине множественного поражения нервов, которые могут спровоцировать нарушение обменных процессов и токсические воздействия. Так миопатии стали подразделяться на невральные амиотрофии, первично-мышечные заболевания и др.
Все детальнее с развитием медицины становится патогенетическая классификация, в основе которой лежит знание о пораженном заболеванием белке. Согласно этой классификации видами миопатии бывают кальпаинопатия, титинопатия и пр. Выявление дефективного белка позволяет сделать выводы о характере его мутации.
Миопатия подлежит разделению на наследственный и приобретённый тип. В анамнезе наследственных типов иногда содержатся относительно четкие данные о наличие этого заболевания у родственников. Примерами миопатий наследственного типа являются дистрофические миопатии, такие, как миопатия Дюшенна, митохондриальные заболевания, а также болезни накопления, наиболее распространена из которых болезнь Помпе.
Различные виды миопатий имеют разный патогенез, что напрямую зависит от того, какой ген в конкретном случае поражен. Так дистрофические миопатии или миодистрофии являются следствием процесса, в котором происходит нарушение синтеза структурных белков миофибрилл. Болезни накопления — результат снижения количества вырабатываемых ферментов и уменьшения их активности. В данном случае рано или поздно пациента начинает беспокоить мышечная слабость и атрофия мышц, что является ключевыми признаками миопатии.
Профилактика
С самого раннего возраста надо приучать ребенка соблюдать при чтении несколько простых правил:
- расстояние от книги до глаз – не меньше 30 см;
- следить за правильной осанкой за столом;
- не читать лежа;
- читать только при достаточном освещении.
Следует позаботиться о соответствии стола (парты) росту ребенка
Надо обратить внимание и на стул: согнутые в коленях ноги под углом 90 градусов должны достать до пола. Свет при чтении, рисовании и письме должен падать всегда слева для правши и справа для левши
Даже в игровой детской комнате должно обеспечиваться хорошее освещение.
Перед началом школьных занятий следует получить консультацию окулиста и уточнить, за какой партой нужно сидеть ребенку, нужна ли ему коррекция зрения.
Сбалансированное питание и периодическое употребление витаминных комплексов для глаз помогут не только в лечении, но и в профилактике близорукости у детей.
Причины врожденной миопатии
Этиология заболевания до конца не изучена. Врожденные миопатии формируются еще в утробе матери и проявляются в неонатальном периоде.
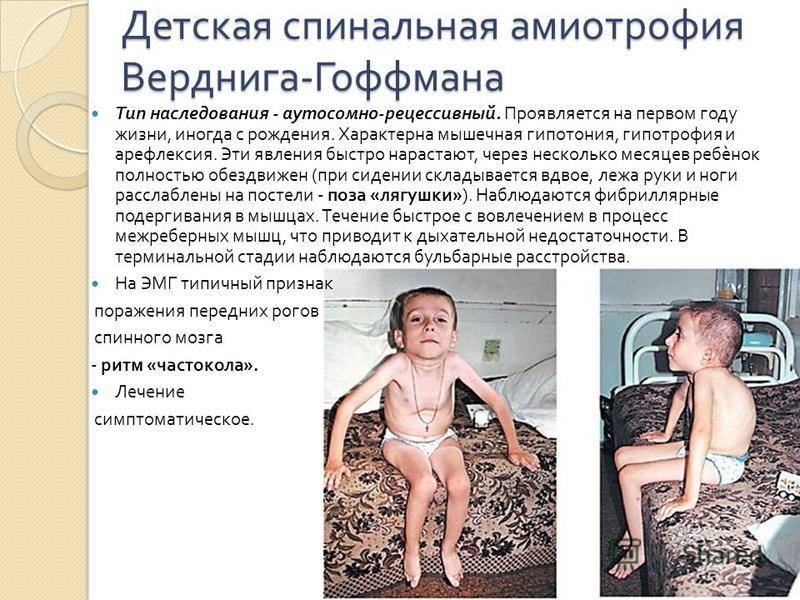
В первые месяцы жизни ребенка отмечается вялость, пассивность, слабое сосание, быстрая утомляемость. После 6 месяцев – затруднения при попытках сидеть, встать на ножки. Малыш отстает от сверстников в физическом развитии. В отдельных случаях обездвиженность наблюдается уже на первом году.







При некоторых видах присоединяются патологии других систем организма:
- психической сферы – задержка психического развития, умственная отсталость;
- сердечно — сосудистой и дыхательной систем – сердечная и дыхательная недостаточность, представляющие угрозу для жизни;
- костной системы – специфическое строение лица (высокое небо), деформации позвоночника (сколиоз, кифоз), врожденный вывих бедра.
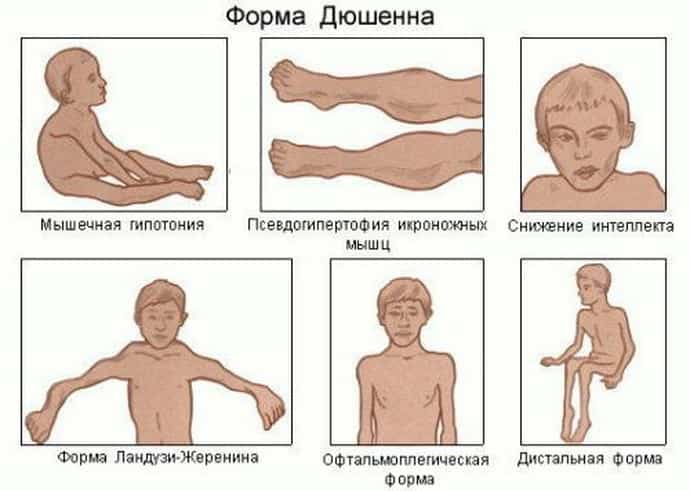
Виды миопатии и ее симптомы
Разновидности заболевания отличаются по типу наследования, локализации измененных генов и виду дефектных белков. Различают 2 патологические группы:
- мышечные дистрофии – дисфункция мышечных волокон, обусловленная дефектом белков, находящихся в их составе;
- структурные миопатии – изменение структуры мышц, вызванное нарушением синтеза белков в период формирования эмбриона.
Классификация врожденных миопатий представлена в таблице.
|
Виды врожденных миопатий |
||
|
Врожденные мышечные дистрофии |
Врожденные структурные дистрофии |
|
|
Мерозин — негативные |
Мерозин — позитивные |
Болезнь центрального стержня |
|
На первый план выходят костные изменения |
Немалиновая |
|
|
С диспропорцией типов мышечных волокон |
||
|
К общим симптомам присоединяется недоразвитие мозга |
Со множественными центральными стержнями |
|
|
Миотубулярная |
||
|
Центронуклеарная |
Остановимся на некоторых формах подробнее.
Болезнь центрального стержня
На первом годужизни ребенок отстает в физическом развитии, наблюдается сниженный тонус мышц. Эти симптомы относят к другим патологиям раннего детского возраста, поэтому диагноз ставят при выраженной клинической картине у более старших детей, когда налицо мышечная слабость, ограничения в движениях и костные деформации. Характерна хрупкость фигуры, маленький рост. Больные могут сохранять некоторую двигательную способность. При этой форме возможно развитие злокачественной гипертермии.
Немалиновая
Тяжелая форма диагностируется во внутриутробном возрасте и после родов грубыми скелетными деформациями, гипотонией мимической мускулатуры и дыхательной недостаточностью, нередко приводящей к смерти.
Легкая форма выявляется после трех лет, даже у подростков, без вышеупомянутых симптомов. В одних случаях болезнь может медленно прогрессировать, в других – вялотекущая.
Выделяют разновидность этой формы, включающей кардиомиопатию, офтальмоплегию, патологию позвоночника.
С диспропорцией типов мышечных волокон
Отмечается генерализованная гипотония, деформации скелета.
Со множественными центральными стержнями
У грудничков наблюдается выраженная мышечная слабость в конечностях. У более старших детей гипотония носит генерализованный характер.


Миотубулярная
Встречается у детей обоего пола, симптомы выражены в легкой степени. Выделена Х-сцепленная разновидность, которая бывает только у мальчиков. Протекает она тяжело, с выраженной атрофией, нарушением глотания и дыхания.
Центронуклеарная
Наиболее распространена разновидность с ранним дебютом, с самого рождения. Характеризуется триадой: дыхательной недостаточностью, гипотонией и нарушениями скелета. Могут присоединяться умственная отсталость и патология глаз. Течение неблагоприятное, ведет к полной инвалидизации.
Подробно о формах и видах миопатии.

Диагностика миопатии
Диагностика врожденных миопатий опирается на биопсию мышечной ткани. При разных видах биопсия показывает:
- отсутствие в мышечных волокнах окислительных ферментов (болезнь центрального стержня);
- палочкоподобные или нитевидные немалиновые тельца (немалиновая миопия);
- множественные мышечные волокна I типа уменьшенного размера в то время, как волокна II типа увеличены или нормального размера (миопия с диспропорцией типов мышечных волокн);
- клетки без митохондрий, с нарушением структуры миофибрилл (миопия со множественными центральными стержнями);
- ядра в мышечных волокнах расположены центрально, что идентично состоянию миофибрилл у 8-10 недельного плода и подтверждает мышечное недоразвитие (миотубулярная).
Остальные методы обследования служат дополнением к постановке диагноза:
- электромиографическое исследование (показывает снижение вольтажа кривой ЭМГ),
- биохимическое исследование (обнаруживает повышение уровня альдолазы и креатинкиназы),
- осмотр врача.
Классификация и формы
Миопатия может быть первичной или вторичной. В первом случае патология развивается в качестве самостоятельного заболевания, во втором — становится последствием прогрессирования других болезней, оказывающих негативное воздействие на соединительную и мышечную ткань ребенка.
В медицинской практике первичная миопатия подразделяется на три отдельные группы — врожденная (симптоматика проявляется в первые месяцы жизни), ранняя детская (патология свойственна детям от пяти до десяти лет) и юношеская форма (проявляется в подростковом возрасте).
По степени выраженности патологического процесса в мышечной ткани миопатия подразделяется на следующие группы:
- Дистальный тип (патологический процесс поражает определенные участки мышц конечностей).
- Проксимальная форма (заболевание поражает мышцы туловища).
- Смешанный тип (сочетает в себе две другие формы заболевания).
Миопатия подразделяется на несколько разновидностей в зависимости от локализации патологического процесса и проявляющейся симптоматики.
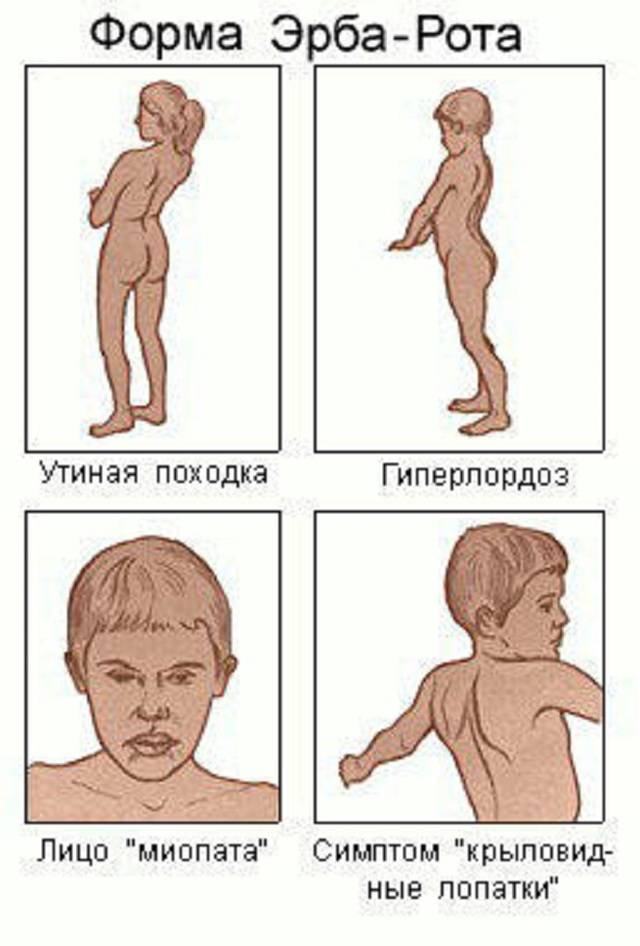
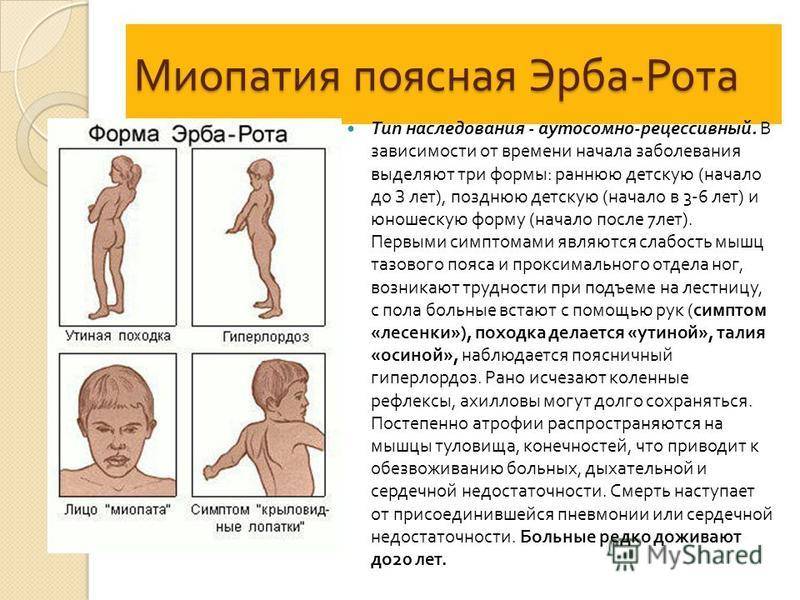
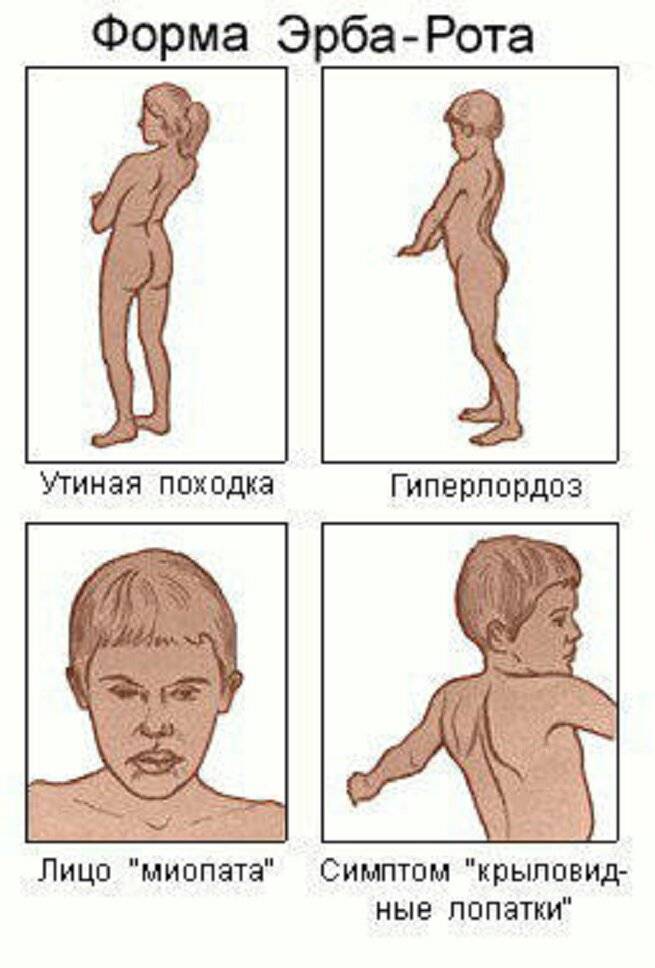
В детском возрасте самыми распространенными формами заболевания считаются синдром Дюшенн-Гризингера, Эрба-Ротта и Ландузи-Дежерина.
В первом случае миопатия является врожденной и поражает мышцы таза. Форма Эрба-Ротта сопровождается поэтапной атрофией мышечной ткани с нижней или верхней части тела. Миопатия Ландузи-Дежерина в большинстве случаев начинается с поражения мышц лица.
Врожденная форма
Врожденная миопатия у детей обусловлена генетическими факторами. Локализация заболевания может наблюдаться в разных локусах хромосом и передаваться на протяжении многих поколений.
Патология приводит к нарушению процесса синтеза определенного вида белка, входящего в структуру мышечной ткани и становится причиной генерализованной слабости мышц ребенка.
В большинстве случаев проявление врожденной миопатии возникает у детей в самом раннем возрасте, а ее симптоматика сохраняется в течение всей жизни.
Причинами врожденной миопатии у детей могут стать следующие факторы:
- митохондриальный дефект;
- недостаточность определенных ферментов, которые участвуют в формировании мышечной ткани;
- нарушение процесса сцепления половых и неполовых хромосом.
Классификацию белково-энергетической недостаточности у детей вы найдете на нашем сайте.
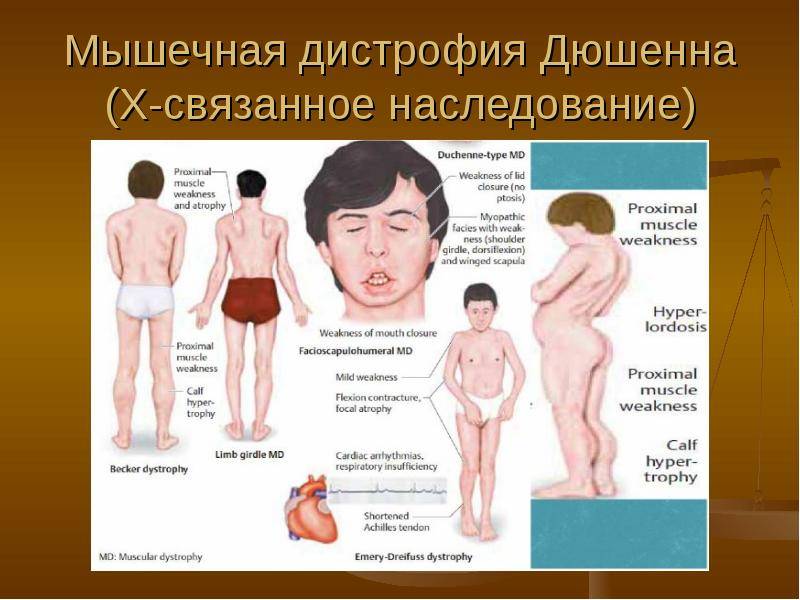
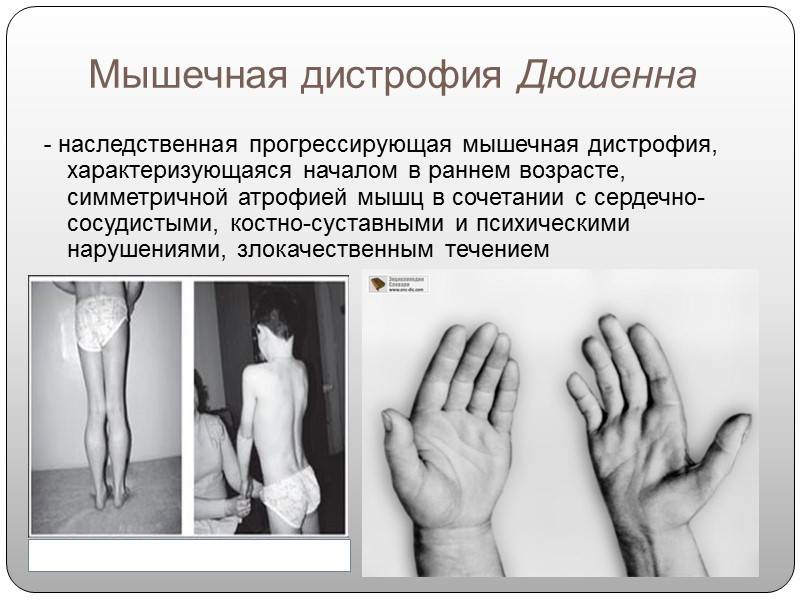
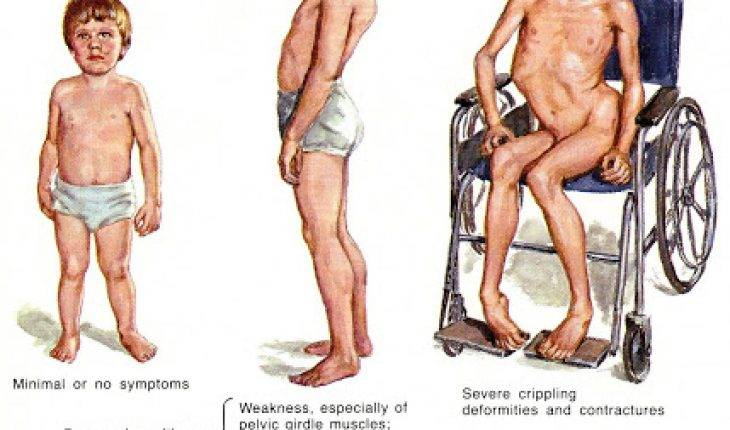
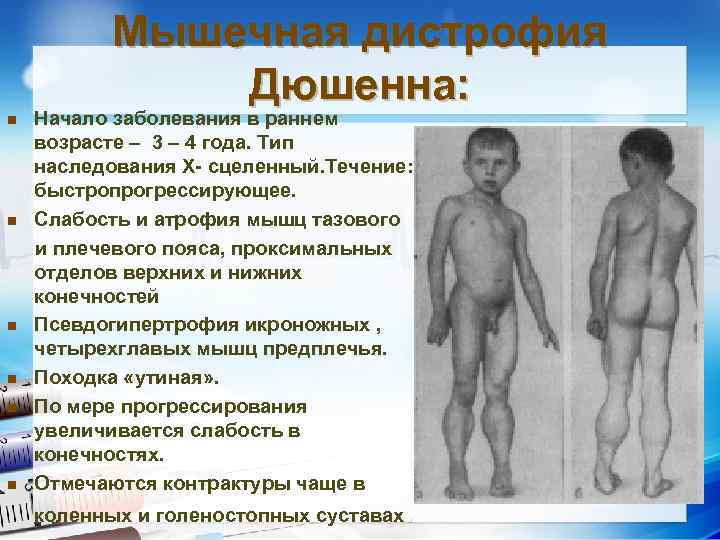
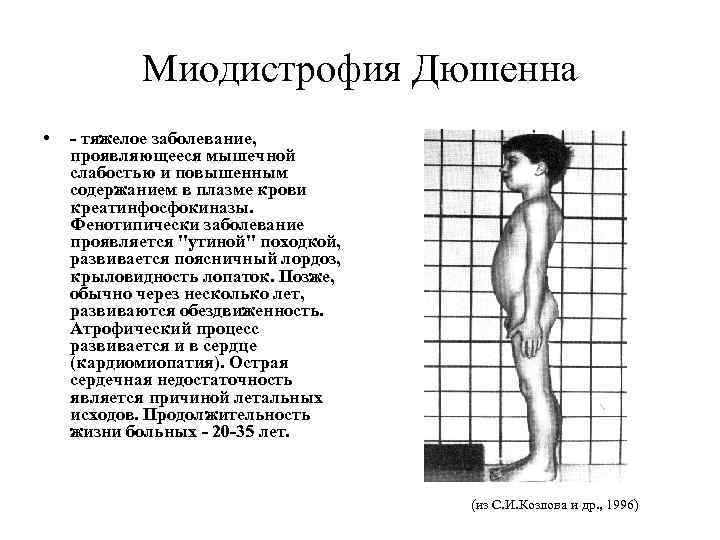
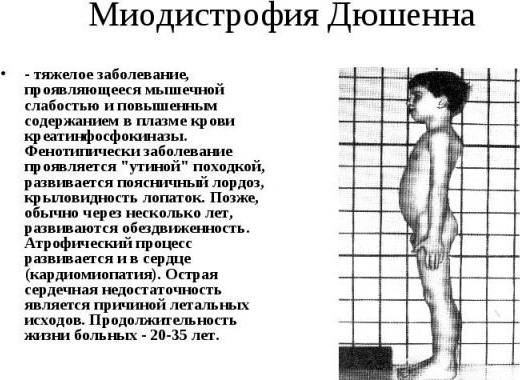
Особенности миопатии Дюшена

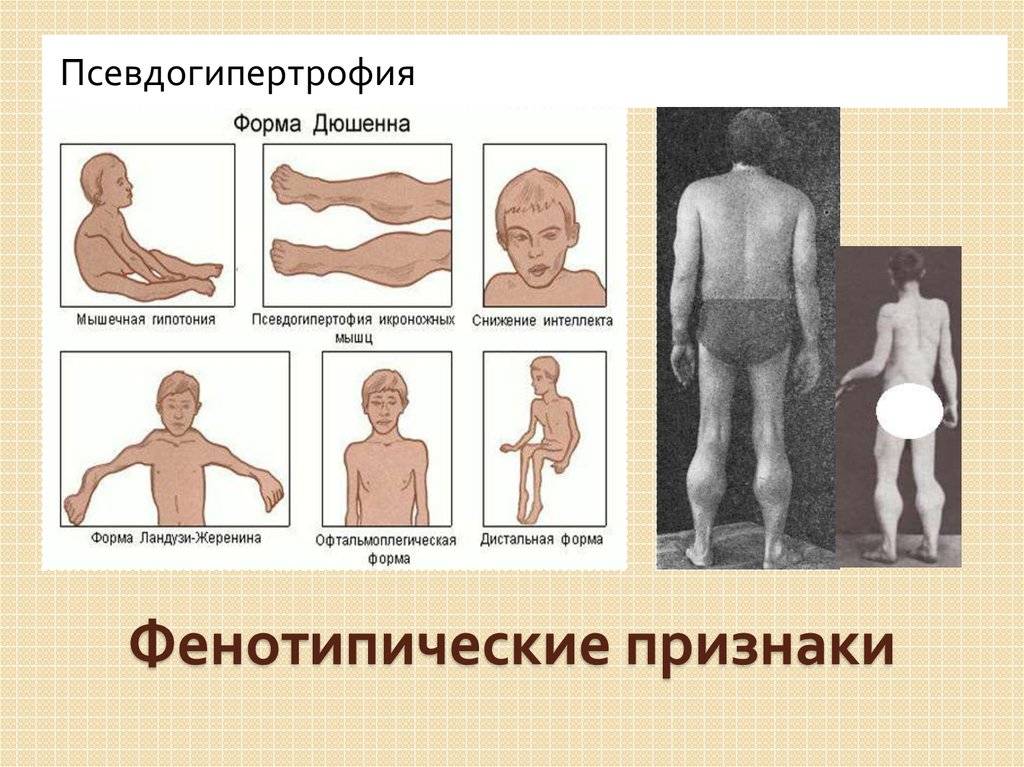
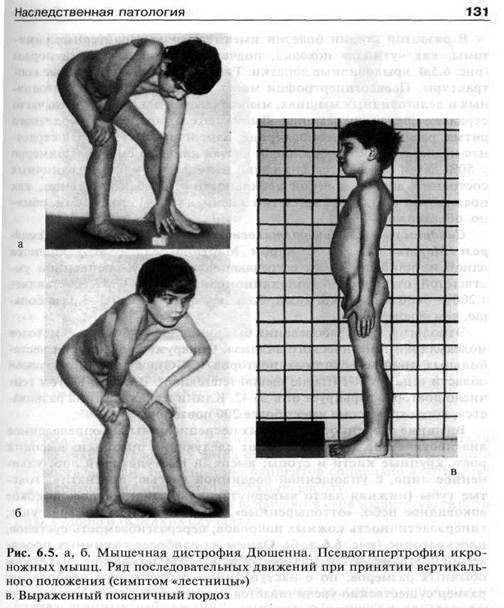
Миопатия Дюшена представляет собой отдельную разновидность заболевания, имеющую псевдогипертрофическую форму.
Симптоматика патологического процесса чаще всего проявляется у детей при достижении трехлетнего или пятилетнего возраста.
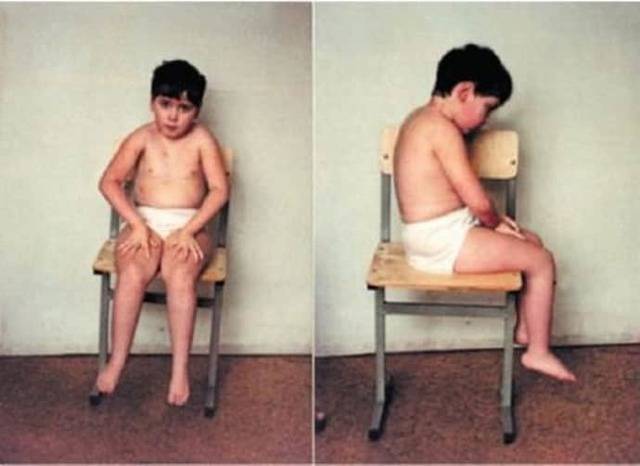
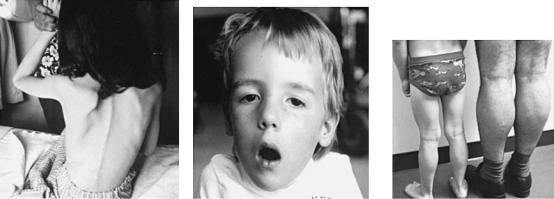
Атрофия мышц начинается с области таза. Падение ребенка может спровоцировать даже малейший толчок. Тело малыша неустойчиво и любые физические нагрузки вызывают у него чувство дискомфорта.
Прогрессирование недуга приводит к полному обездвиживанию ребенка.
Особенности патологического процесса:
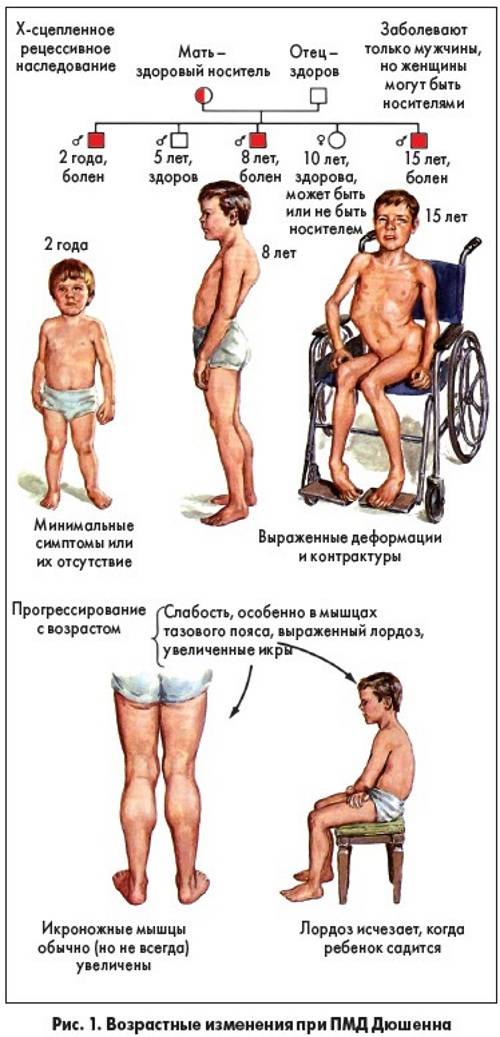
- данная форма миопатии диагностируется только у мальчиков;
- болезнь всегда начинает развиваться с поражения мышц таза;
- постепенно атрофия распространяется на область плеч и спины;
- патологический процесс сопровождается сколиозом, сердечной и дыхательной недостаточностью;
- мышцы верхних конечностей атрофируются на последнем этапе развития недуга;
- осложнением миопатии такого типа может стать олигофрения.
Лечение
Основная задача в лечении пациентов с наследственными миопатиями — отсрочить наступление обездвиженности с быстрым формированием контрактур и дыхательных расстройств.
- Немедикаментозное лечение.
ЛФК.
Пассивные и активные движения в суставах по несколько раз в день. Дыхательная гимнастика. Интенсивность нагрузки зависит от стадии болезни и носит щадяще-охранительный режим, чтобы не спровоцировать ухудшение состояния.





- Массаж в щадящем режиме.
- Ортопедическая коррекция направлена на предупреждение развития патологических установок в руках и ногах, борьбу с контрактурами с помощью специальных ортопедических шин, укладок для пациента.
- Диета с большим содержанием белка, витаминов и микроэлементов.
- Медикаментозное лечение.
Возможности лекарственной помощи значительно ограничены, так как нет специфического лечения. Симптоматическое лечение направлено на поддержание активности здоровой мышечной ткани, уменьшение контрактур и атрофий.
Лечение включает в себя следующие группы препаратов:
- Витамины группы В, витамины А и Е.
- Нестероидные анаболики (оротат калия, АТФ).
- Кардиотрофические препараты (рибоксин, карнитина хлорид).
- Корректоры микроциркуляции (пентоксифиллин).
- Ноотропы (пирацетам).
- Хирургическое лечение также направлено на борьбу с контрактурами при неэффективности консервативных методов. Производят рассечение сухожилий или мышц (ахиллотомия, миотомия).
Лечение других миопатий ведется в рамках заболевания, которое их вызвало (лечение гриппа, токсоплазмоза и др.).
Первый медицинский канал, врач-невролог Левицкий Г. Н. читает лекцию на тему «Приобретенные и метаболические миопатии»:
Образовательная программа по неврологии, выпуск на тему «Миопатии»:
Осложнения и последствия
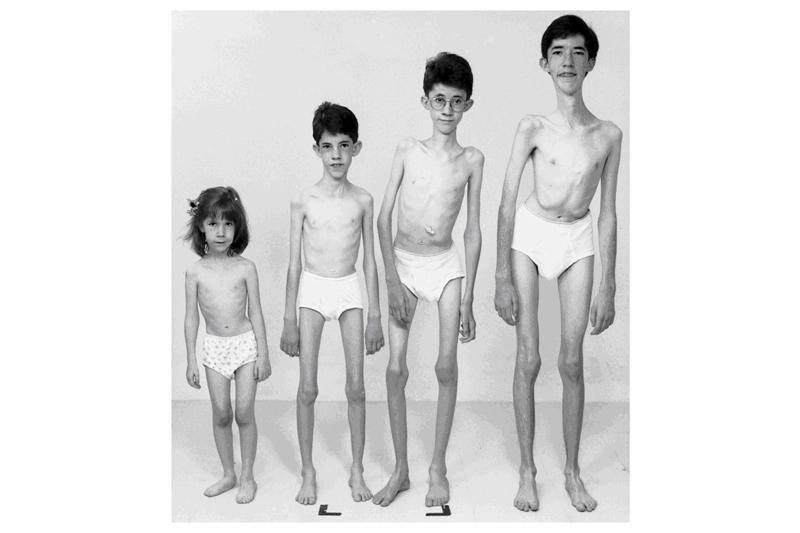
Как уже говорилось ранее, данное заболевание сильно укорачивает жизнь человека и это можно считать самым серьезным последствием. Так как патология быстро прогрессирует, то мышцы становятся слабыми, возникают необратимые последствия в работе сердца, наблюдается сбой дыхательной системы.
Если миопатия Дюшена будет своевременно выявлена и лечение будет подобрано правильно, то пациент сможет дожить до 30 лет.
К осложнениям можно отнести такие заболевания и патологии:
- Остеопороз. Чтобы постараться отсрочить данную патологии рекомендуется принимать витамин Д и кальций, поэтому стоит скорректировать питание. При остеопорозе принимают биосфосфонаты.
- Заболевания суставов и позвоночника. Суставы становятся практически полностью подвижными, поэтому рекомендуется носить шины. В некоторых случаях проводят операции.
- Заболевания пищеварительной системы и проблемы с весом. С возрастом человек начинает терять в весе из-за некроза мышц, поэтому требуется постоянная консультация диетолога. Также пациента может потревожить и запор, который также является следствием малоподвижного образа жизни. В данной ситуации рекомендуется принимать слабительные препараты, больше есть продуктов, которые богаты на клетчатку. В возрасте от 17 лет у больного могут возникнуть проблемы с жевательной и глотательной функцией. В редких случаях проводиться гатростомия.
- Нарушения дыхательной функции. Из-за слабого кашлевого механизма могут прогрессировать респираторные инфекции, так как бактерии и слизь не выводятся. В качестве профилактики рекомендуется делать прививки. Также по мере прогрессирования патологии у пациента наблюдается снижение уровня кислорода в организме, как следствие возникают утренние головные боли, бессонница, слабость, нервная возбудимость, особенно во сне.
- Заболевания сердца. В большинстве случаев развивается кардиомиопатия.
Профилактика болезни затрудненная, так как данная патология является генетической, поэтому нужно уделить внимание консультации у генетиков тем семьям, у которых наблюдается отягощенная наследственность. Статья написана по материалам сайтов: www.dnalab.ru, meduniver.com, neurodoc.ru
Статья написана по материалам сайтов: www.dnalab.ru, meduniver.com, neurodoc.ru.
Диагностика
Возможно антенатальное выявление патологии по результатам исследования околоплодных вод (амниоцентеза).
Диагностика семейных форм и типичных случаев миопатии обычно не представляет сложности. Клиническое обследование выявляет мышечную слабость, как правило, изменяющуюся в течение дня. Пациента просят надуть щеки, вытянуть губы, зажмуриться, а затем быстро открыть глаза (вызывает затруднение). Проверяют наличие двоения в глазах, дизартрии, дисфагии. Исследуется сила подвздошно-поясничных и дельтовидных мышц, мышц сгибателей и разгибателей шеи (сгибатели более слабые, чем разгибатели).
В сомнительных случаях прибегают к следующим методам диагностических исследований:
- игольчатая электромиография – определяется миопатический паттерн, для которого характерна сниженная амплитуда М-ответа, при этом усиленная интерференция и полифазность потенциала;
- магниторезонансная томография (МРТ) мышц – определяется жировое и/или соединительнотканное перерождение мышечных волокон;
- биопсия мышц – позволяет подтвердить диагноз и дифференцировать ряд миопатий;
- исследование креатин-креатининового обмена – для миопатии характерно наличие креатинурии, повышение уровня креатинкиназы в сыворотке крови (особенно при псевдогипертрофической миопатии);
- генетическое исследование, включая молекулярно-генетический анализ, генетическое тестирование членов семьи, особенно способных к деторождению.
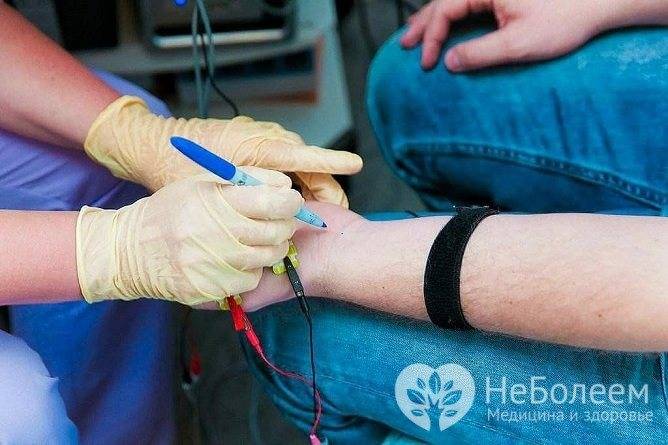
Электромиография — метод, используемый в диагностике миопатий
При приобретенных миопатиях диагностический поиск зависит от первопричины, например, при тиреоидной форме проводят определение уровня гормонов щитовидной железы.
Дифференциальная диагностика
Врожденные миопатии у детей следует дифференцировать с атонико-астатической формой детского церебрального паралича (ДЦП) и диспластическими кифосколиозами неясной этиологии.
Наследственные миопатии
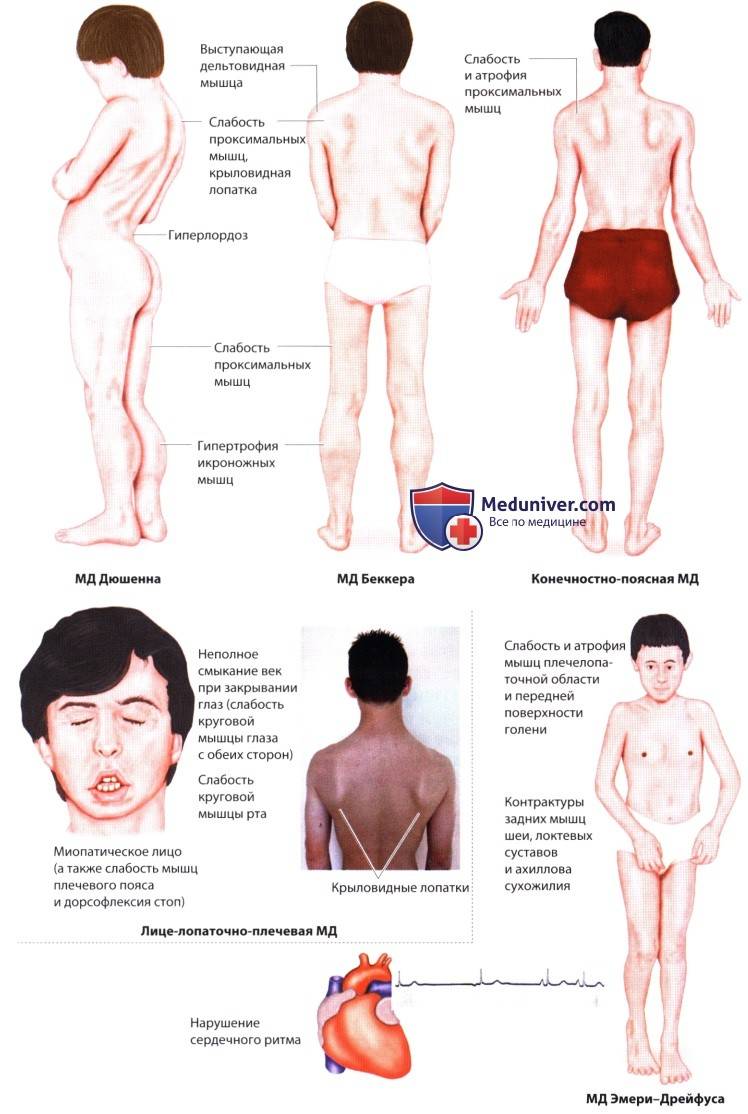
Примером болезней, протекающих с симптомами наследственных миопатий – детская псевдогипертрофическая мышечная дистрофия Дюшенна, поздняя дистрофия Беккера, плечелопаточная дистрофия Ландузи-Дежерина, и другие, более редкие формы: лопаточно – перонеальная форма Давиденкова, ювенильная дистрофия Эрба, офтальмоплегическая форма миопатии, дистальная миопатия Веландера, а также ряд врожденных и непрогрессирующих форм. Рассмотрим симптомы наиболее часто встречающихся миопатий.
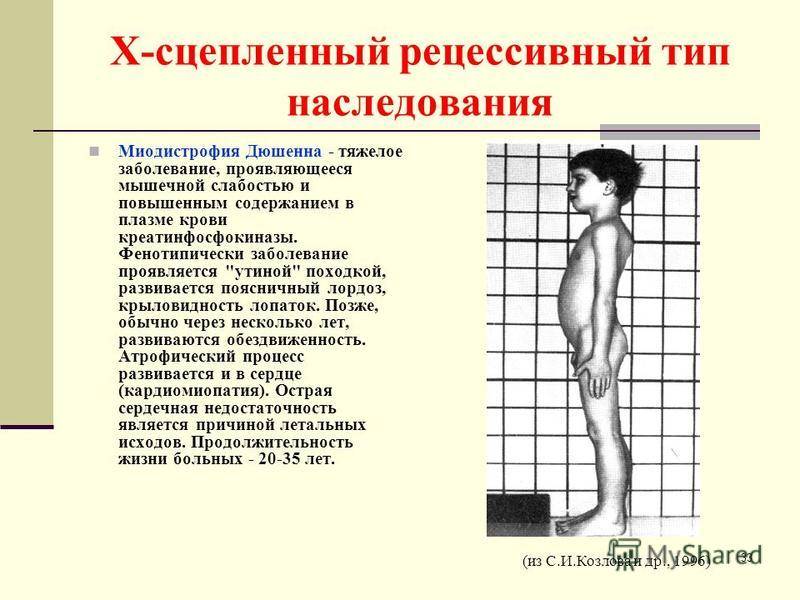
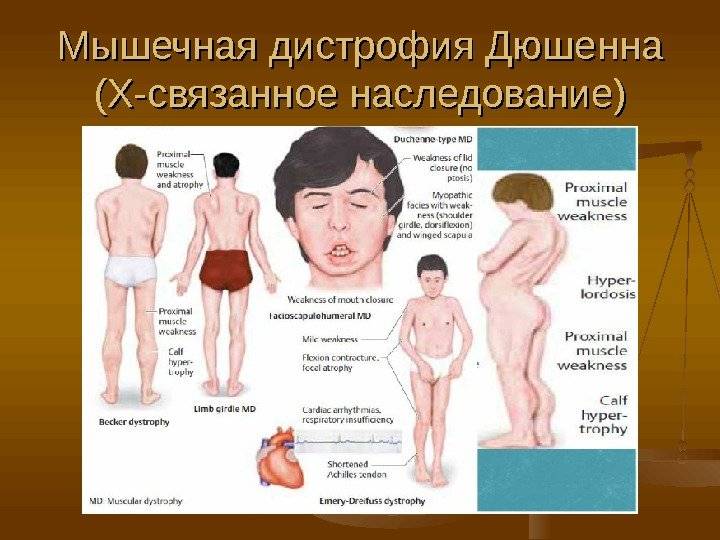
Детская миодистрофия Дюшенна
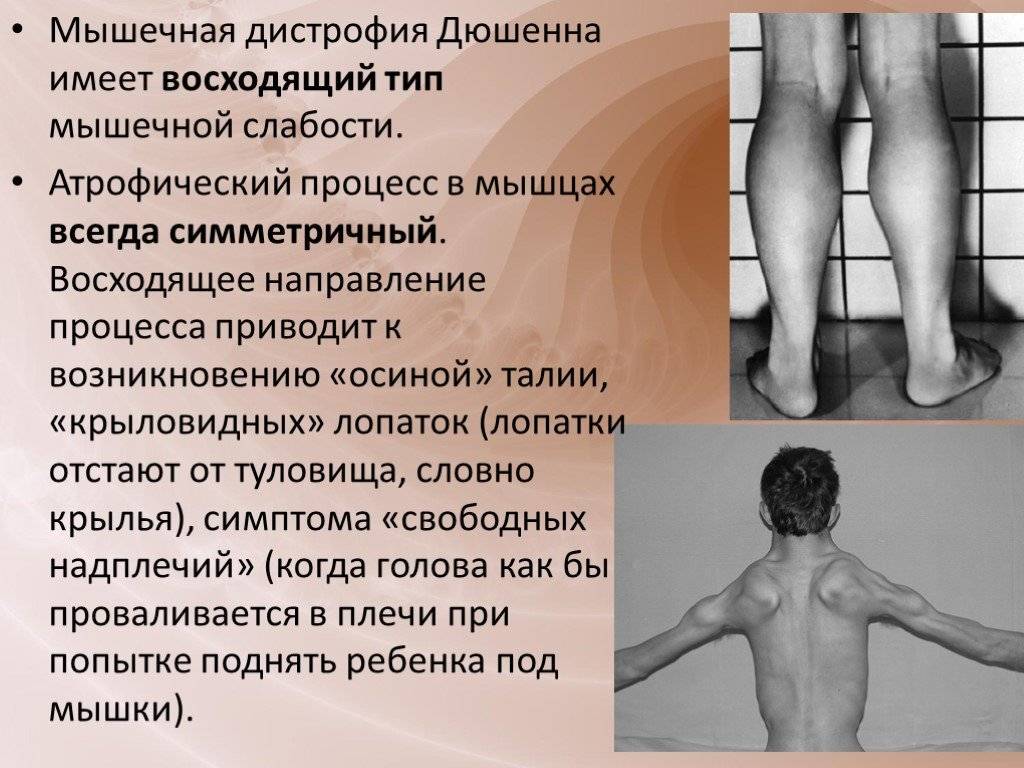
Является злокачественной дистрофией, и встречается довольно часто – 1 случай на 3,5 тысячи, и только у мальчиков. Частота не зависит от расы, географии, социального положения. Болезнь проявляется прогрессированием мышечной слабости. Кроме этого, миопатия приобретает признаки мышечной гипотрофии, а затем – и атрофии.
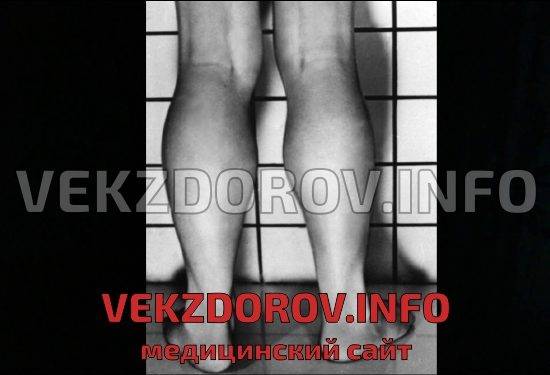
Болезнь начинается рано, мышечная слабость возникает вначале в районе мышц таза, затем плеч, в возрасте ребенка до 3 лет.
Симптомы
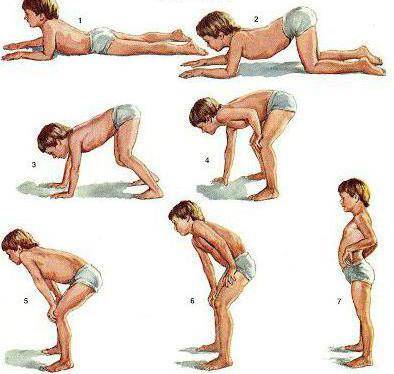
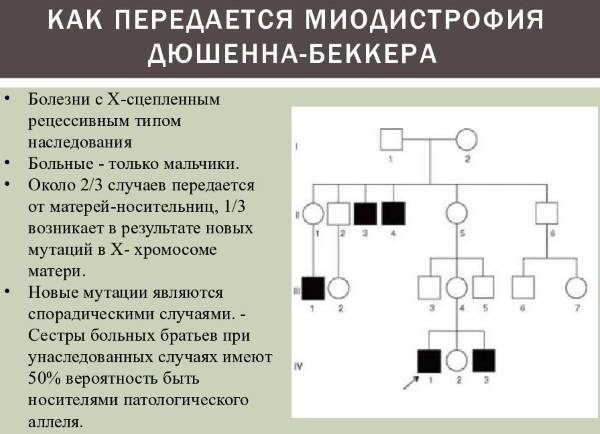
При этой наследственной миопатии прогноз неблагоприятный: развивается полная обездвижннность, и в возрасте не старше 20 лет возникает смертельный исход. У больных детей часто возникает умственная отсталость, и недоразвитие интеллекта. Высокая частота встречаемости этого заболевания связана с тем, что белок «дистрофин», который находится на коротком плече «женской» Х-хромосомы, имеет очень высокую способность к возникновению спонтанных мутаций.

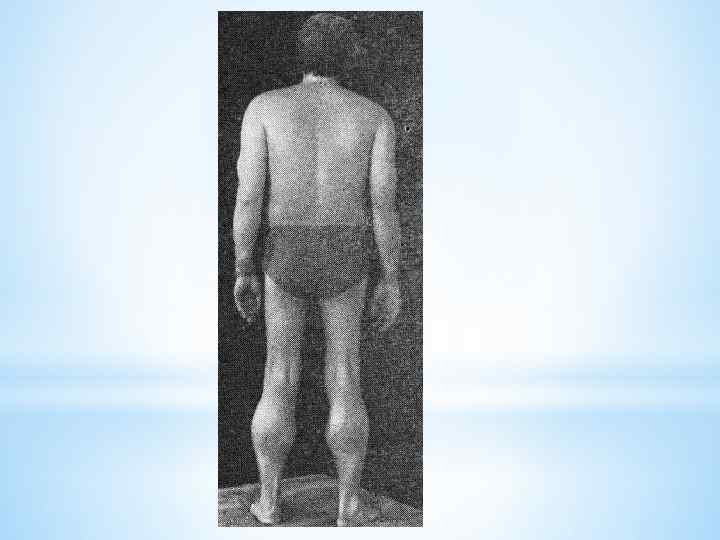
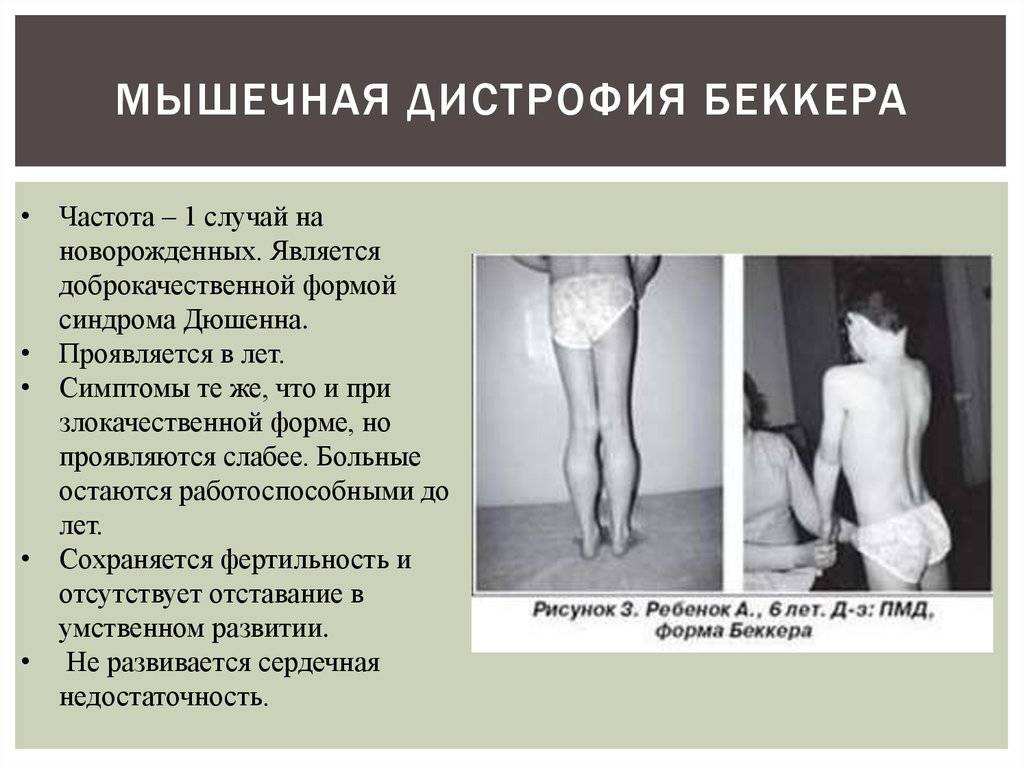




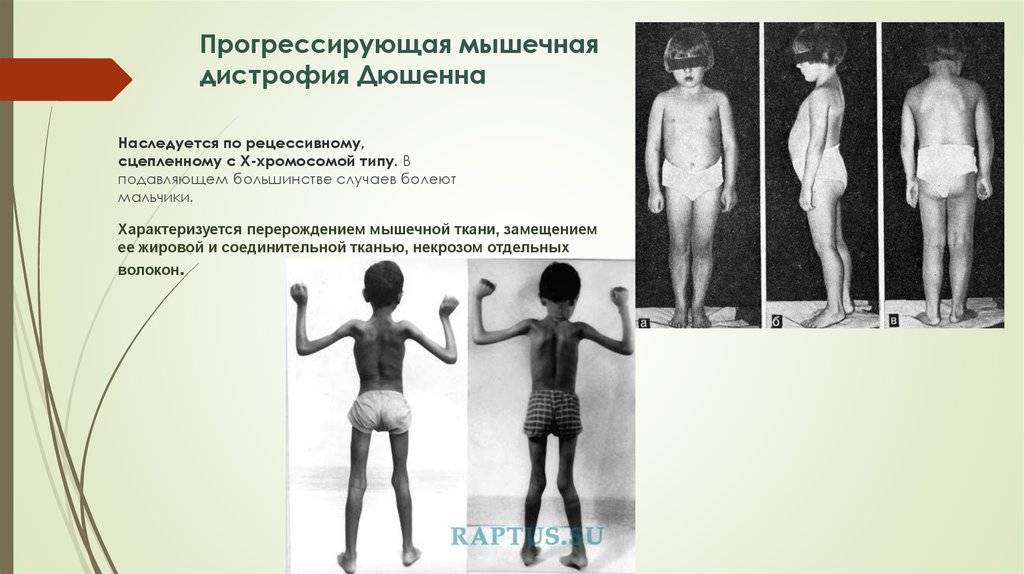
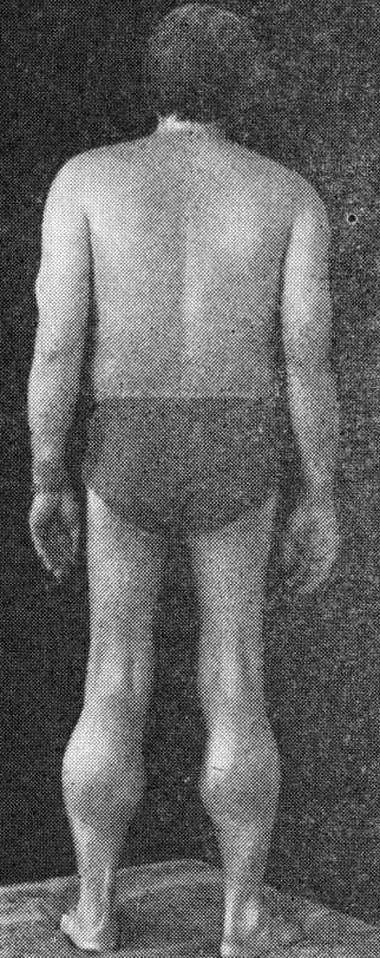
Поздняя псевдогипертрофическая миодистрофия Беккера
По клинике это заболевание протекает так же, как и болезнь Дюшенна. Но при этом оно начинается после 10-летнего возраста, возникает в 8 раз реже, чем предыдущее заболевание.
Заболевание также кодируется в виде рецессивного типа, сцепленного с полом, и для диагностики миодистрофей Беккера и Дюшенна можно применять одни и те же пробы ДНК.
Миопатия Ландузи – Дежерина
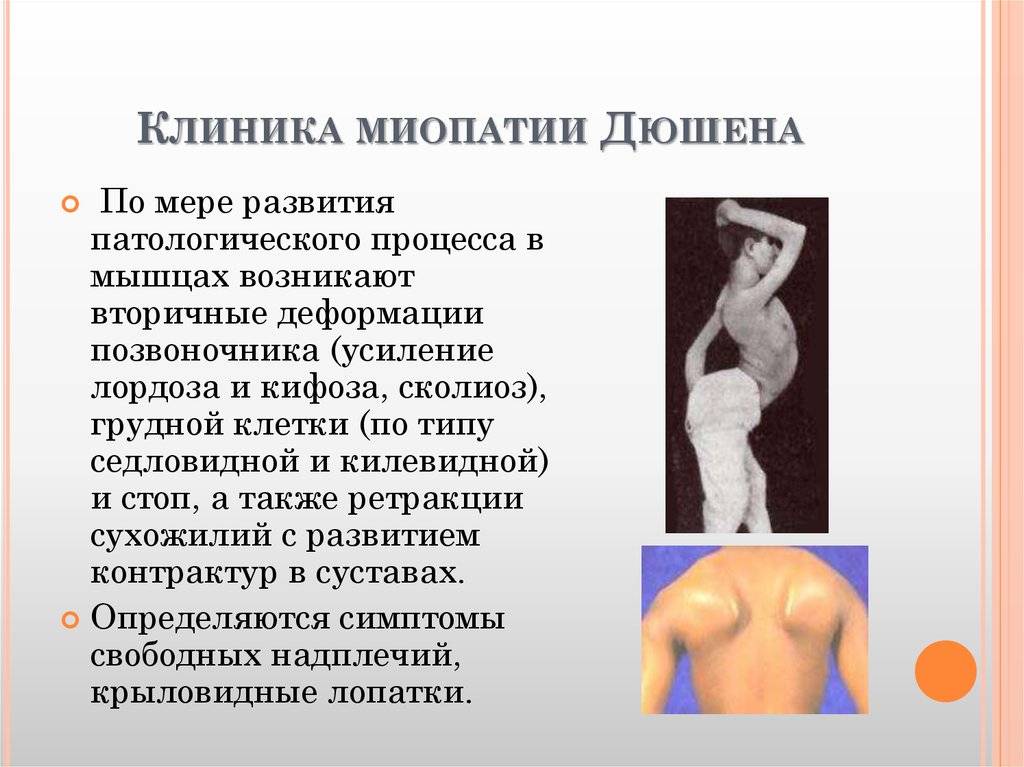
Это более редкий вид миопатии. Ее еще называют плечелопаточно-лицевой, по той причине, что поражаются мышцы лица и плечевого пояса. Мимика снижена, веки полностью не смыкаются, круговая мышца рта гипертрофирована, возникает симптом «губ тапира». Возникает гипотрофия, атрофия и парезы в проксимальных отделах рук, появляется такой симптом, как «крыловидные лопатки». При далеко зашедшем течении возникает поражение мышц ног и тазового пояса. В таком случае пациент с миопатией Ландузи – Дежерина может быть оформлен на получение пособия по инвалидности.

Это заболевание связано с тем, что фермент КФК (креатинфосфокиназа) не может превращать креатин в креатинфосфат, связывая его с АТФ. В результате мышца не получает высокоэнергетического соединения, которое может использоваться при повышенной мышечной работе. В результате накапливается КФК в мышцах, происходит их атрофия, а в поздних случаях – денервация.
Лечение при миопатии Ландузи – Дежерина симптоматическое. Прогноз – для жизни благоприятный, для качества жизни – возможно инвалидизация, начиная с возраста в 30-40 лет. При всех наследственных миопатиях специфического лечения не разработано. Применение гормонов приводит к замедлению прогрессирования поражения мышц, при выраженной дистрофии мышц показано хирургическое лечение (например, операция артродеза) при привычном вывихе болтающегося сустава.

Этиология и патогенез миопатий
Развитие миопатии чаще всего обусловлено наследственными причинами. Генетические виды этого заболевания имеют разные формы, для которых характерны определенные типы наследования. Важную роль в этиологии миопатий играют острые и хронические инфекции, травмы и повреждения, алиментарные дистрофии и прочие неблагоприятные факторы окружающей среды. Специалисты также указывают на перенапряжение, которое может вызвать развитие миопатии у людей, предрасположенных к ней ввиду наследственности.
Таким образом, толчком к проявлению и развитию миопатий может стать практически любой неблагоприятный фактор человеческой жизни. Природу предрасположенности некоторых пациентов к развитию миопатий позволили распознать биохимические и генетические исследования. Точно известно, что у больных наблюдаются дефекты метаболизма, обусловленные наследственностью. Они провоцируют нарушение функции определенных ферментов, что становится причиной недостатка содержания в кровяной сыворотке альдолазы, трансаминазы, креатинфосфокиназы и других жизненно важных веществ.
В патогенезе миопатий, помимо генетических аномалий обмена разной степени, выявлено нарушение симпатической иннервации мышц. Поражение затрагивает преимущественно проксимальный отдел рук и ног, где она богаче, чем в других зонах. Страдают от миопатий и центральные вегетативные аппараты диэнцефального уровня. Это проявляется нарушением некоторых центрально обусловленных вегетативных рефлексов.
Лечение: цели, методы, трудности
Лечение миопатии Дюшенна невозможно, можно только продлить время жизни и поддержать состояние пациента на стабильном уровне.
Различные методы современной медицины помогают облегчить состояние, замедлить прогрессирование болезни. Ниже будут представлено лечение миодистрофии Дюшена для разных возрастов, но бывают такие случаи, когда требуется применять сразу несколько способов терапии:
- Пациент в возрасте до 5 лет. В данной ситуации не требуется радикальное лечение. Рекомендуется осведомить родителей о заболевании, рассказать о проблемах и последствиях, рассказать, какие нагрузки допустимы для ребенка, и провести в обязательном порядке генетическую консультацию.
- Пациент в возрасте до 8 лет. Большинству детей в таком возрасте уже требуется поддержка мускулатуры нижних конечностей. Кортикостероиды помогают замедлить ход развития заболевания на некоторое время. Рекомендуется принимать Преднизолон или Дефлазакорт.
- Пациент в возрасте от 8 лет до 20 лет. Наблюдается постепенное ослабление мышц, которое в результате заставляет ребенка прибегнуть к инвалидному креслу.
- Пациент в возрасте от 20 лет. Препараты в данной ситуации помогут только облегчить состояние, начинает прогрессировать инфекция дыхательных путей.
В течение всей жизни пациент должен принимать препараты, которые обеспечивают поддержку организма:
- витамины В и Е, аминокислоты, кальций, анаболические гормоны, калия оротат;
- Прозерином, Галантаминон, Оксазилон.
Также курсами проводят инъекции Ретаболила, АТФ, Цереброзилил, Анарилин. Рекомендуется принимать глютаминовую кислоту, Ексазил.
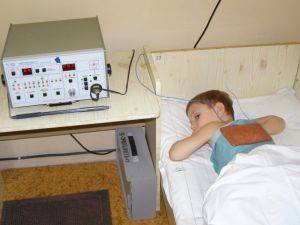 Параллельно проводиться лечение ЛФК, которое осуществляется непродолжительными курсами с перерывами. Также рекомендуется делать массаж, электрофорез Прозерина, Липазы, кальция хлорида, применяют лечебные ванны и индуктотермию. В особо тяжелых случаях лечение проводиться в домашних условиях. Чтобы продлить жизнь на несколько лет можно попробовать глюкокортикоидное лечение.
Параллельно проводиться лечение ЛФК, которое осуществляется непродолжительными курсами с перерывами. Также рекомендуется делать массаж, электрофорез Прозерина, Липазы, кальция хлорида, применяют лечебные ванны и индуктотермию. В особо тяжелых случаях лечение проводиться в домашних условиях. Чтобы продлить жизнь на несколько лет можно попробовать глюкокортикоидное лечение.
Параллельно пациент должен постоянно находиться под наблюдением кардиолога. Также обязательно ребенок должен полноценно питаться. В рацион включены: растительные жиры и белковая пища, свежие или приготовленные на пару овощи, фрукты, молочные продукты, овсянка, яйца, мед, орехи и морковь. Не стоит потреблять крепкий чай или кофе, спиртные напитки, приправы, сахар, капусту и картофель.