Признаки и симптомы
Часто болезнь проявляет себя до 3 месяцев и всегда до 1–2 лет. Выявляются симптомы выраженной анемии:
- бледность кожи (зачастую этим и исчерпывается);
- младенец быстро устает сосать грудь и начинает задыхаться; сонливость;
- общая слабость;
- сниженный аппетит;
- вялые мышцы;
- непереносимость физических нагрузок и задержки физического развития;
- диспепсические явления.
У трети больных сопутствуют различные аномалии развития:
- малый вес при рождении;
- широко расставленные глаза;
- выпуклый лоб;
- седловидный нос;
- аномалии челюстно-лицевой части черепа (волчья пасть);
- короткая шея;
- гипогонадизм;
- полифалангия большого пальца на руках, олигодактилия – пальцев на руке меньше нормы;
- урологическая патология.
Реже встречаются пороки сердца и аномалии скелета. Течение хроническое. Симптомы могут быть различной степени по своей выраженности, что зависит от показателей крови, правильности лечения и длительности патологии.
Симптомы анемии Даймонда-Блекфена
На первый план при синдроме Даймонда-Блекфена выступают анемические симптомы – бледность, слабость ребенка, у грудных младенцев часто развивается гипотрофия, наблюдается недобор массы. Примерно у половины больных помимо нарушений со стороны крови также возникает ряд физических отклонений – микроцефалия, гипертелоризм, птоз век, микрогнатия. Возможны аномалии скелета – увеличение размера лопаток и кистей, отсутствие некоторых пальцев, задержка роста костной ткани. В некоторых случаях возможны такие нарушения как «заячья губа». Поражаются и органы зрения – развивается косоглазие, глаукома, катаракта. Многие указанные симптомы возникают в раннем возрасте ребенка и усугубляются выраженной анемией, поэтому своевременно начатое лечение может значительно ослабить или даже устранить многие из них.
В отличие от транзиторных и приобретенных анемий, синдром Даймонда-Блекфера незначительно влияет на работу печени и селезенки – их заметное увеличение может возникать лишь на конечных стадиях заболевания или в результате осложнений гемотрансфузионной терапии.
Дополнительная информация, влияющая на течение и исход заболевания
В целом прогноз для жизни достаточно благоприятный. Выполнение данных клинических рекомендаций позволяет сохранить полноценную работоспособность пациента. Продолжительность жизни ограничена в первую очередь развитием осложнений от проводимой терапии.
Спонтанная ремиссия АДБ возможна в примерно 20% случаев к 25 годам независимо от ранее проводимой терапии .
Осложнение заместительной терапии эритроцитной массой – посттрансфузионная перегрузка железом – может существенно сокращать продолжительность жизни и ухудшать качество жизни больных .
Продолжительность жизни больных: до 40 лет доживает 75,1±4,8% больных; в случае достижения ремиссии или медикаментозной ремиссии выживаемость составляет 85-100%; трансфузионно зависимые пациенты доживают до взрослого возраста в 60% случаев .
Общая выживаемость после родственной совместимой ТГСК, если она проводилась до 9-летнего возраста, составляет 95%, после неродственной полностью совместимой ТГСК – 85% .
Смертность пациентов с АДБ зависит от развития и степени тяжести осложнений от проводимой терапии (посттрансфузионная перегрузка железом, инфекции, осложнения после ТГСК) – 67%, связана с прогрессией заболевания (тяжелая аплазия кроветворения, злокачественные заболевания) – 22%, не установлена причинная связь – 11% случаев .
При отсутствии ТГСК избегать профессий, связанных возможными травмами. При успешной ТГСК ограничений в выборе профессии нет.




Без ТГСК детородная функция обычно не страдает, после проведенной ТГСК возможно бесплодие.
Физиологические изменения, происходящие при беременности, могут вызвать повышение потребности как в ГКС, так и в трансфузиях эритроцитной массы. Во время беременности может быть возврат клинических проявлений и необходимость терапии у больных со спонтанной компенсацией АДБ.
Пренатальная диагностика и генетическое консультирование:
При генетическом консультировании необходимо учитывать высокую вероятность рождения больного ребенка в данной семье при последующих беременностях. Пренатальная диагностика возможна при идентифицированной мутации у пациента; в
30% случаев мутацию выявить не удается, следовательно проведение пренатальной диагностики становиться невозможно.
При генетическом консультировании необходимо учитывать крайне высокую вероятность рождения больного ребенка.
У пациентов мужского пола при идентифицированной мутации возможно проведение генетического исследования спермы для определения риска передачи данного заболевания следующему поколению. В случае выявления только мутантного аллеля – риск рождения больного ребенка составляет 100%, деторождение не рекомендуется. В случае наличия нормального и мутантного аллеля – показана ЭКО с предимплантационной диагностикой.
У пациентов женского пола при идентифицированной мутации возможно ЭКО с предимплантационной диагностикой.
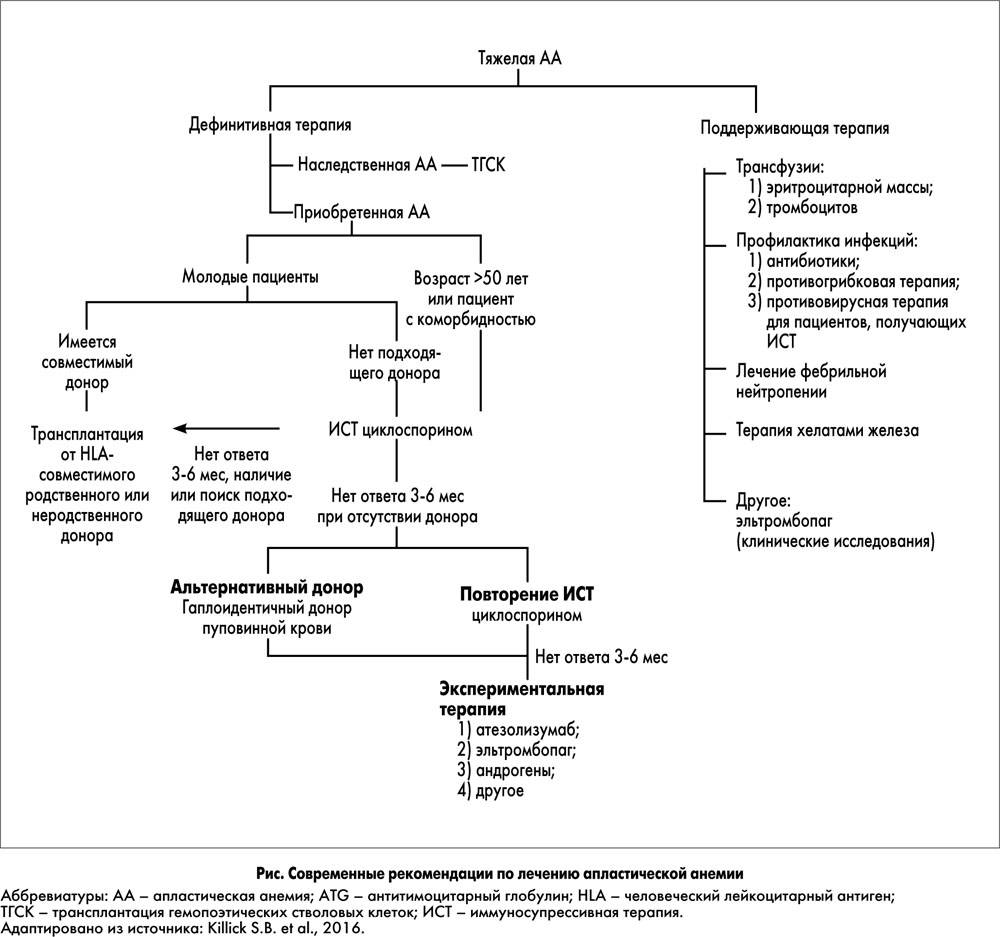
Лечение
Основной метод лечения – глюкокортикостероиды в течение длительного времени. Но на них реагируют только 70–80% больных. У таких пациентов терапия дает улучшение уже через 2 недели: ретикулоцитарная реакция выражается в увеличении ретикулоцитов (молодые эритроциты) в крови и увеличивается гемоглобин до 90–100 г/л. Далее доза гормона корригируется до минимально эффективной. Такие больные наиболее благоприятны в прогностическом плане.
У некоторых пациентов отмечается задержка во времени ответной реакции на кортикостероиды либо нестойкая ремиссия. Они получают курс кортикостероидов повторно.
Около 15% больных остаются резистентными к действию гормонов. Им лечение проводится гемотрансфузиями – эритроцитарной массы и приемом хелаторов железа (они связывают и выводят избыток железа из организма).








Пожалуй, единственным шансом на полное излечение является сегодня аллогенная трансплантация (пересадка от донора) костного мозга. Однако процедура нередко дает осложнения, поэтому ее применяют лишь при неэффективности гормонов.
Заболевание протекает волнообразно, с периодическими ремиссиями. В этом состоянии лечение пациентам не проводится.
1.3 Эпидемиология
По данным Kynaston et al (1993) расчетная частота встречаемости заболевания составляет 1 на 100000 или 1 на 200000 рожденных живыми детей , по данным других авторов – 5-7 на 1000000 рожденных живыми детей вне зависимости от национальности и пола 25. По данным Российского регистра в ежегодно в стране рождается 8-11 детей с АДБ, что в среднем составляет 4,975 случаев на 1000000 рожденных живыми детей. Около 45% больных – семейные случаи с аутосомно-доминантным путем наследования, оставшиеся 55% больных – спорадические случаи .
Кумулятивный риск развития всех злокачественных новообразований у больных АБД превышает общепопуляционный в 5,4 раза. Максимальный риск развития был отмечен для миелодиспластического синдрома, острого миелобластного лейкоза, аденокарциномы толстой кишки, остеогенной саркомы и злокачественных опухолей женских половых органов .
Не менее 40% пациентов с АДБ нуждаются в проведении постоянной трансфузионной терапии . Около 75% пациентов с АДБ доживают до возраста 40 лет, для трансфузионно-зависимых пациентов этот показатель составляет чуть более 57% .
1.2 Этиология и патогенез
В настоящее время данные о генетических нарушениях (повреждение рибосом за счет нарушения формирования их субъединиц), лежащих в основе патогенеза АДБ, приводящих к гаплонедостаточности рибосом, позволяют отнести данное заболевание к группе рибосомопатий . Мутации в генах рибосомальных белков приводят к нарушению синтеза как малых, так и больших субъединиц рибосом, что индуцирует р53 с последующим арестом клеточного цикла на границе фаз G1/S 8, было показано, что данные изменения не затрагивают другие клеточные линии . При этом известно, что мутации в различных генах рибосомальных белков по-разному влияют на дифференцировку клеток эритроидного ряда . Например, мутации в гене RPS19 индуцировали снижение пролиферации клеток-предшественников, однако конечная дифференцировка эритроцитов оставалась ненарушенной. В то же время мутации в гене RPL11 приводили не только к резкому подавлению пролиферации эритроидных предшественников, но и к торможению дифференцировки эритроцитов и значительному увеличению апоптоза в культуре клеток .
Еще одним объяснением нарушения эритропоэза может служить тот факт, что эритроидная дифференцировка сопровождается разительной перестройкой ядерных структур с конденсацией хроматина, являющейся подготовительным шагом для утраты ядра. Вследствие этого ядрышко подвергается структурным и молекулярным изменениям, что может потенцировать рибосомальный стресс, вызванный мутациями в рибосомальных белках, и вести к апоптозу . В норме транскрипты будущих рибосомальных белков образуются в ядре РНК-полимеразой II, транслируются в цитоплазме, после чего данные белки транспортируются в ядрышко, где принимают участие в формировании рибосом. 40S и 60S субъединицы рибосом затем эспортируются из ядрышка через нуклеоплазму в цитоплазму, где соединяются в 80S субъединицей рибосомы и выполняют свою роль в синтезе белка в клетке .
По некоторым данным, мутации в генах RPL5 и RPL11 чаще ассоциируются с наличием врожденных аномалий, чем мутации в гене RPS19, причем первые характеризуются более тяжелым фенотипом .
В настоящее описан большой спектр мутаций и делеций различных генов рибосомальных белков, наиболее часто встречаемые поломки в генах RPS19, RPS10, RPS24, RPS26, RPL5, RPL11, RPL35a, RPS7, RPS17 19
Идентифицированы также единичные случаи АДБ в результате мутации генов GATA1, FLVCR1 и TFR2 .
2.2 Физикальное обследование
Общий осмотр подразумевает оценку общего физического состояния, роста и массы тела, наличия вторичных половых признаков в соответствующем возрасте., выявление врожденных аномалий развития. Более чем у половины больных АДБ выявляются врожденные аномалии развития. Пороки развития, кроме низкого роста, встречаются в 47% случаев: аномалии черепа и лицевого скелета (гипертелоризм, высокий выпуклый лоб, готическое небо, небная расщелина, плоская спинка носа, микрогнатия, микроцефалия, микротия, низко расположенные ушные раковины) – 50%, и аномалии кистей рук (удвоенный, расщепленный, 3-фаланговый большой палец, синдактилия) – 38%, патология сердца (дефект межжелудочковой перегородки, дефект межпредсердной перегородки, коарктация аорты, тетрада Фалло) – 30%, и мочеполовой системы (подковообразная почка, удвоение мочевыводящих путей, гипоспадия – 39%, сочетанные пороки развития встречаются в 21% случаев .










Физическое развитие низкое. Низкий вес при рождении встречается в 10% случаев, при этом в половине из этих случаев отмечается отставание физического развития от гестационного возраста. Более 60% больных имеют рост менее 25 перцентиля .
Оценивая причину низкого роста у пациентов АДБ трудно отделить конституциональные особенности от побочных эффектов проводимой терапии (перегрузка железом вследствие постоянных гемотрансфузий или длительный прием глюкокортикостероидов) .
2.1 Жалобы и анамнез
Основная жалоба – бледность кожи и слизистых, слабостью, утомляемостью (у детей первых месяцев жизни проявляется быстрым утомлением при кормлении, особенно грудью матери) . В дальнейшем (у детей старше 1 года) присоединяются жалобы на отставание физического роста ребенка .
Сбор анамнеза при АДБ подразумевает тщательный расспрос о возрасте появления первых симптомов заболевания, наличие в семье детей или взрослых с аналогичными проявлениями (заболеванием) .
Средний возраст начала клинических проявлений – 2 месяца жизни, средний возраст установления диагноза – 3-4 месяца . В более 90% случаев манифестация заболевания на первом году жизни, крайне редко – в первые сутки жизни .
Причины анемии Даймонда-Блекфена
Непосредственной причиной четверти случаев анемии Даймонда-Блекфена служит мутация в гене RPS19 расположенном в 19-й хромосоме, который кодирует важный рибосомальный белок S19. Последний входит в состав малой (40S) субъединицы рибосомы человека. Такая мутация наследуется по аутосомно-доминантному механизму со встречаемостью 6 случаев на миллион человек. В других случаях были обнаружены мутации иных генов, однако они так или иначе связаны с рибосомальными белками – это гены RPS7, RPS24, RPL5, RPL32A и ряд других. Распространенность таких мутаций, характер их наследования, доля в общем количестве заболевших анемией Даймонда-Блекфена, их влияние на прогноз и исход патологии в настоящий момент остается объектом изучения врачей-генетиков. Также интерес представляет собой вопрос, почему мутации генов рибосомальных белков оказывают влияние именно на эритропоэз и почти не влияют на другие ростки кроветворения.
В процессе изучения находится и патогенез этого заболевания. Существует несколько теорий, пытающихся объяснить торможение образования эритроцитов в красном костном мозге. Наиболее распространенные указывают в качестве возможных причин анемии Даймонда-Блекфена дефекты микроокружения клеток-предшественников эритроцитов, их внутренние аномалии, супрессию со стороны иммунной системы или гуморальные факторы, останавливающие созревание эритробластов. Ни одна из теорий на сегодняшний день не получила достоверного и однозначного подтверждения.
При этом заболевании в красном костном мозге наблюдается неуклонное снижение эритроидных единиц, причем примерно в трети случаев данный процесс начинается во время внутриутробного развития, что позволяет диагностировать анемию Даймонда-Блекфена сразу после рождения. Соответственно, начинает снижаться количество выделяемых в кровь эритроцитов, в костном мозге накапливаются эритробласты, что может вводить в заблуждение (подобные изменения характерны для лейкоза). При этом у младенцев уровень фетального гемоглобина может не снижаться, поэтому данный показатель не считается диагностическим в случае анемии Даймонда-Блекфена. Возникает компенсаторный рост уровня эритропоэтинов в крови, однако они в данном случае не способны увеличить скорость образования эритроцитов. В конечном итоге развивается выраженная анемия.
2.3 Лабораторная диагностика
- • Нормохромная, обычно макроцитарная, анемия в раннем возрасте без вовлечения других клеточных линий.
- • Ретикулоцитопения.
- • Нормоклеточный костный мозг с селективным уменьшением эритроидных предшественников ( 100 г/л, нормальное число ретикулоцитов;
частичный — Hb 85-100 г/л, наличие ретикулоцитов;
отсутствие ответа — Hb 500 мкг/л (уровень убедительности доказательства В) . Отменяться хелаторная терапия может при достижении верхней границы возрастной нормы содержания ферритина сыворотки при условии прекращения заместительной трансфузионной терапии и нормализации содержания железа в печени и миокарде, оцененных методом МРТ Т2* (для печени возможно методом определения содержания железа в сухом веществе печени). Хелаторы: деферазирокс (начальная доза 30 м г/кг/сут peros ежедневно, далее с шагом 5 м г/кг/сут повышается до максимальной дозы 45 м г/кг/сут или понижается в зависимости от концентрации ферритина сыворотки) (уровень убедительности доказательства В) , при содержании ферритина сыворотки менее 500 мкг/л доза снижается до 125-250 мг/сут (уровень убедительности доказательства С) , дефероксамин (начальная доза 40 мг/кг/сут подкожно 5 дней в неделю в виде длительной инфузии (8-12 ч), при необходимости интенсивной хелации, в случае развития застойной сердечной недостаточности 100 мг/кг/сут непрерывно внутривенно капельно в течение 7-10 дней (уровень убедительности доказательства В) ). Для интенсификации хелаторной терапии может использоваться комбинация деферазирокса (30 мг/кг/сут per os ежедневно) в сочетании с дефероксамином (40-50 мг/кг/сут подкожно ежедневно в течение 8-12 ч) (уровень убедительности доказательства B) . Применение деферипрона в качестве препарата 1-й линии нецелесообразно в связи с высоким риском развития агранулоцитоза, и его назначение возможно только при наличии противопоказаний к деферазироксу и дефероксамину.
При проведении хелаторной терапии необходимо контролировать:
сывороточное железо, ОЖСС/НЖСС, НТЖ, сывороточный ферритин каждые 3 мес при подборе дозы хелатора, далее каждые 6 мес;
клиренс эндогенного креатинина до начала хелаторной терапии, каждые 3 мес на этапе подбора дозы, далее каждые 6-12 мес;
МРТ Т2* печени и миокарда 1 раз в год.
3.3 Хирургическое лечение
При данной патологии не используется.
3.4 Иное лечение
Трансплантация гемопоэтических стволовых клеток рассматривается в качестве радикальной терапии .
При отсутствии эффекта на ГКС-терапию трансплантация гемопоэтических стволовых клеток (ТГСК) от родственного или неродственного HLA-совместимого донора рекомендуется рассматривать как альтернатива пожизненной заместительной терапии эритроцитной массой для пациентов младше 9 лет .
Уровень убедительности доказательства В
ТГСК рекомендуется рассматривать как радикальный метод лечения для пациентов младше 9 лет в случае наличия родственного HLA-совместимого донора у трансфузионно зависимых пациентов, не отвечающих на глюкокортикоиды и учитывая риск прогрессивного угнетения кроветворения и развития злокачественных заболеваний у больных АДБ, ответивших на ГКС-терапию .
уровень убедительности доказательства В
Комментарий: В настоящее время, по данным регистров АДБ Франции и Германии, бессобытийная выживаемость при проведении ТГСК от родственного HLA-совместимого донора в возрасте младше 9 лет составляет 94%, а в более старшем возрасте 55%. При этом родственный донор должен быть обследован для исключения субклинической формы АДБ .








У пациентов младше 9 лет неродственную HLA-совестимую ТГСК рекомендовано рассматривать как вариант радикальной терапии
Уровень убедительности доказательства С .
Комментарий: В настоящее время, по данным регистров АДБ Франции и Германии, бессобытийная выживаемость при проведении ТГСК от неродственного HLA-совместимого донора пациента младше 9 лет составляет 85% .
Уровень убедительности доказательства D
Комментарий: Описаны случаи достижения ремиссии при использовании лейцина (по 800 мг/кг/м2 три раза в сутки), однако в клинических рандомизированных исследованиях эффективность такой терапии не доказана .глюкокортикостероидная терапия