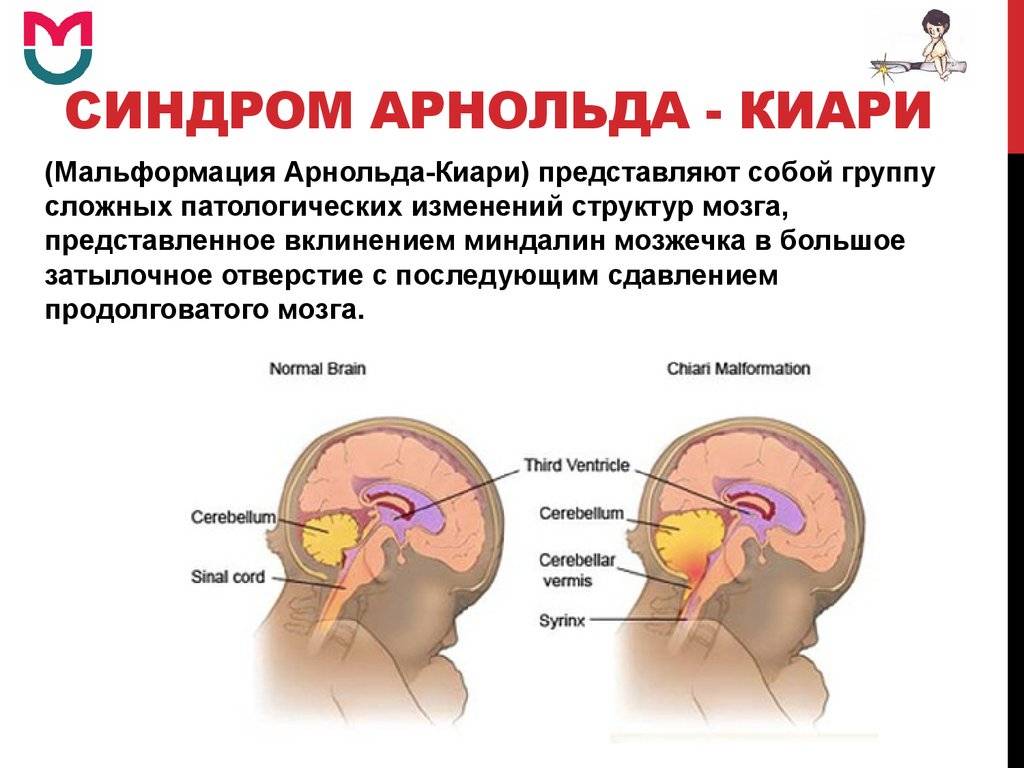
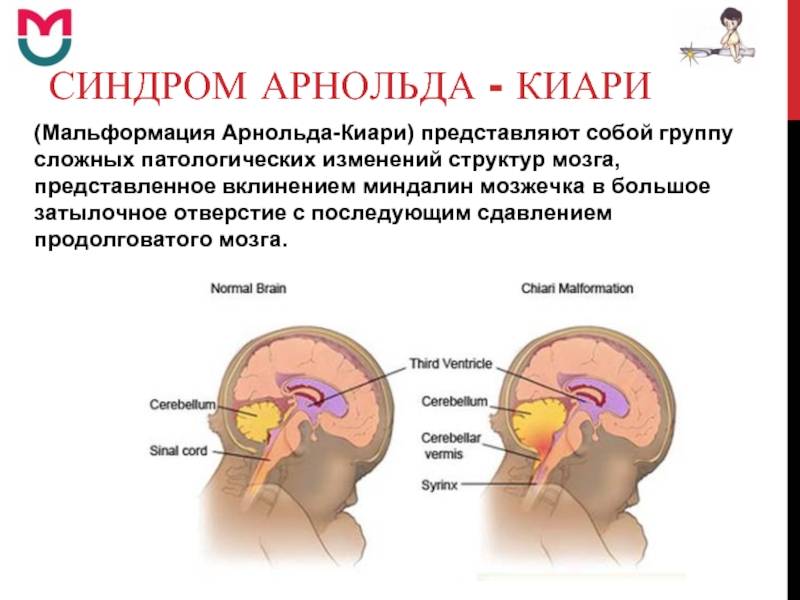
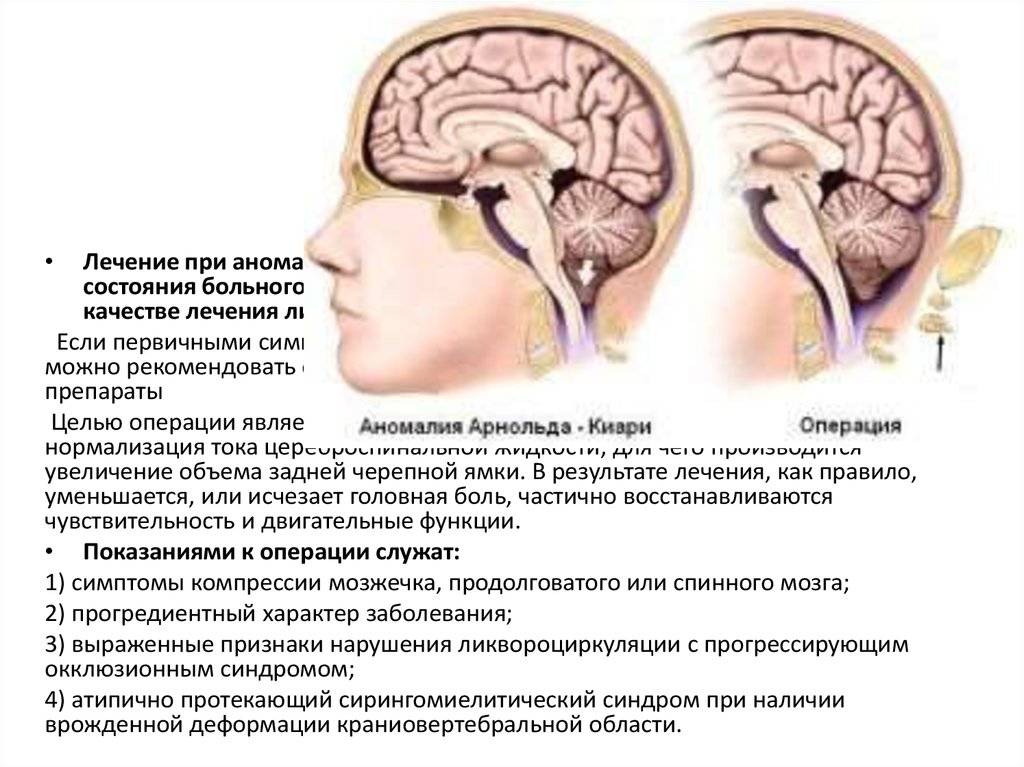
Что такое синдром Арнольда-Киари?
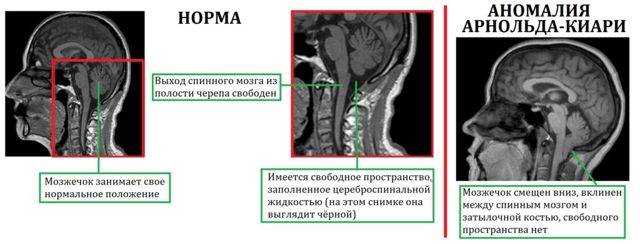
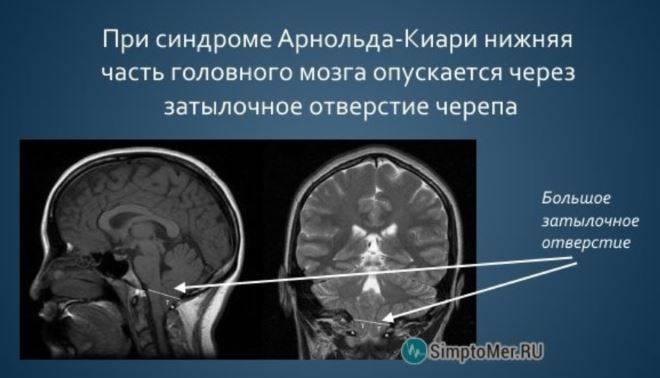
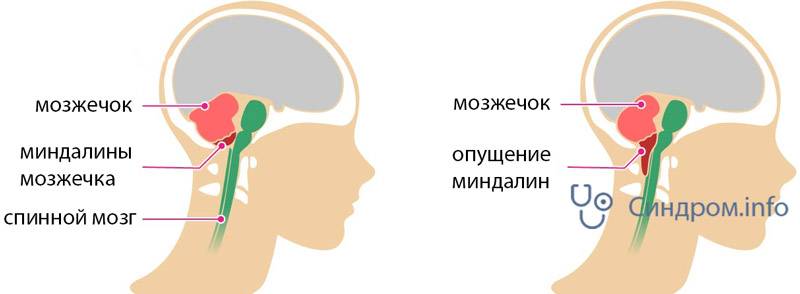
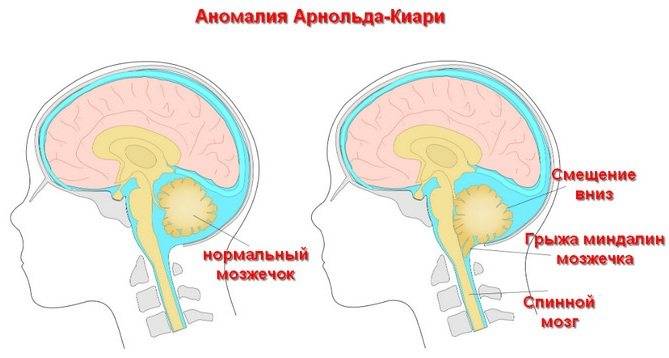
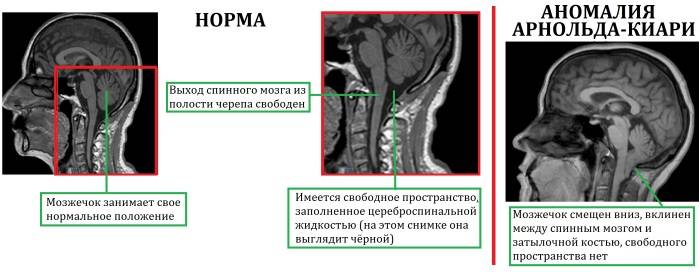
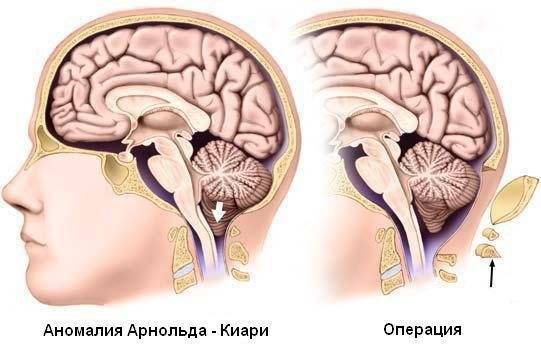
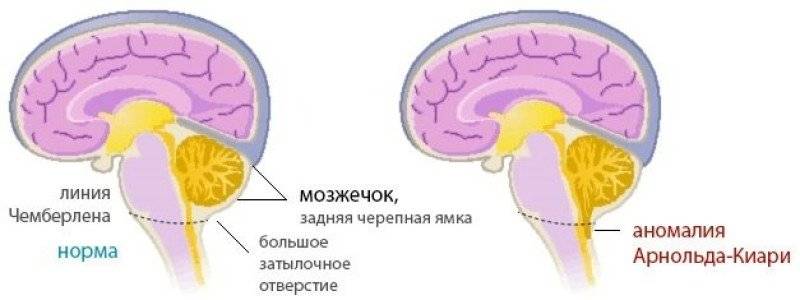
При такой аномалии вся конструкция задней ямки черепа опускается ниже большого затылочного отверстия.
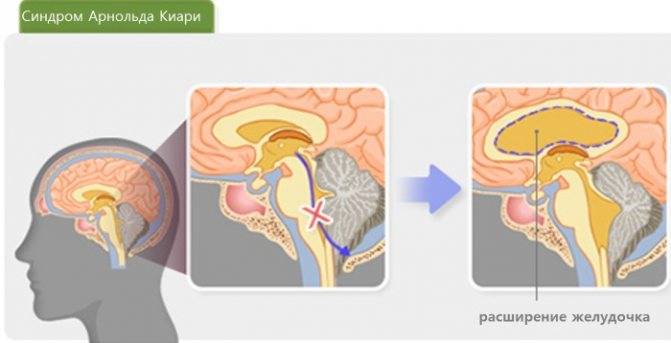
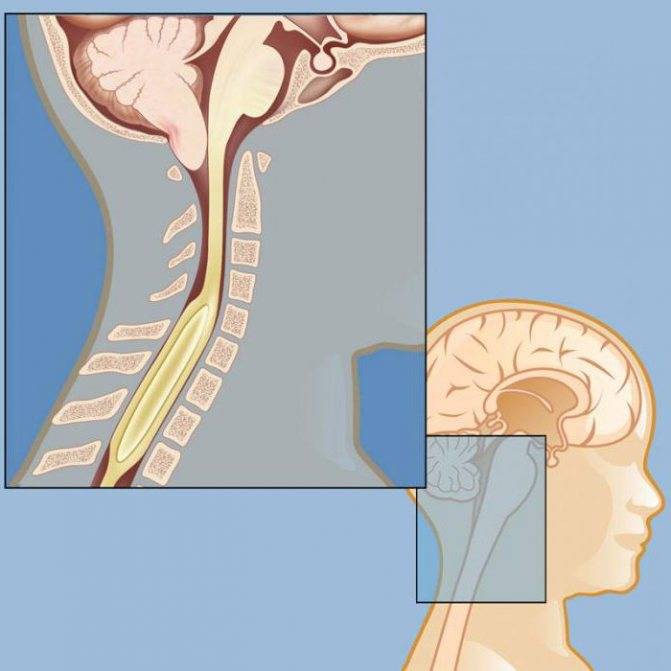
БЗО является пограничной зоной между спинным и головным мозгом. При отсутствии патологии ликвор или спинномозговая жидкость свободно двигается в субарахноидальном пространстве. При синдроме Арнольда-Киари миндалины мозжечка закупоривают отверстие, тем самым нарушая отток ликвора, что способствует образованию гидроцефалии.
Синдром Арнольда-Киари имеет три типа – I,II и III, IV. Все они связаны с заниженным расположением миндалин мозжечка.
При общей патологии, типы МК имеют определенные отличия:
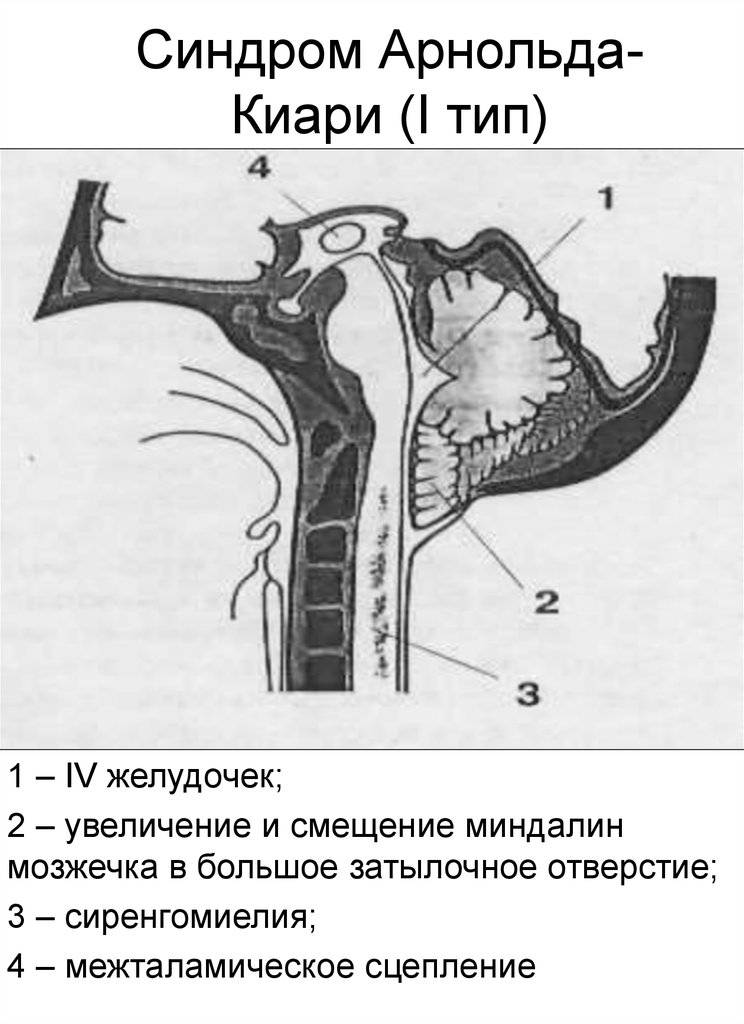
I тип синдрома представляет собой опущение миндалик и смещение малого мозга ниже БЗО. При этом виде аномалии задняя черепная ямка имеет небольшой размер.
При проведении диагностики можно выделить три варианта синдрома – передний, промежуточный и задний:
- При переднем варианте наблюдается смещение С2 назад, свисание над зубовидным отростком, базилярная импрессия и платибазия;
- При наличии промежуточного варианта происходит сжатие сместившимися миндалинами мозжечка вентрального отдела продолговатого мозга и верхних сегментов спинного;
- Задний вариант приводит к компрессии верхнешейных секций спинного и дорсальных отделов продолговатого мозга.
Первый вид мальформации Киари неврологами считается самым распространенным заболеванием. Чаще других им страдают женщины, поздно рожавшие и вступившие в предклимактерический период.
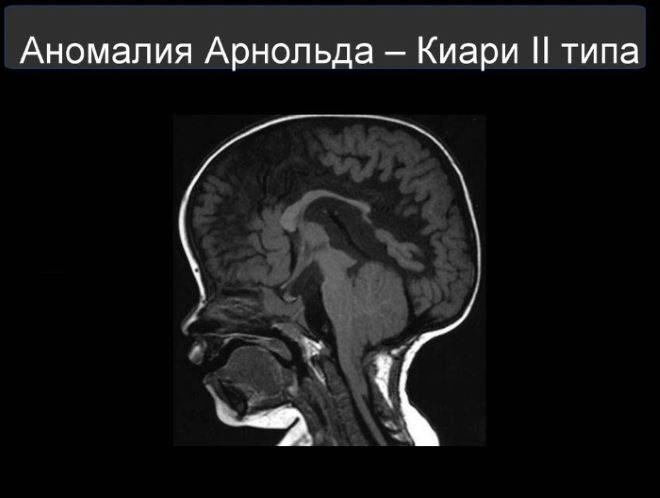
- II тип синдрома имеет врожденную форму. Прогрессирование патологии происходит во время внутриутробного развития. Аномалию можно выявить при обследовании плода. Продолговатый мозг, IV желудочек, нижний отдел червя вклиниваются на 2 мм ниже входа в большое заднее отверстие.
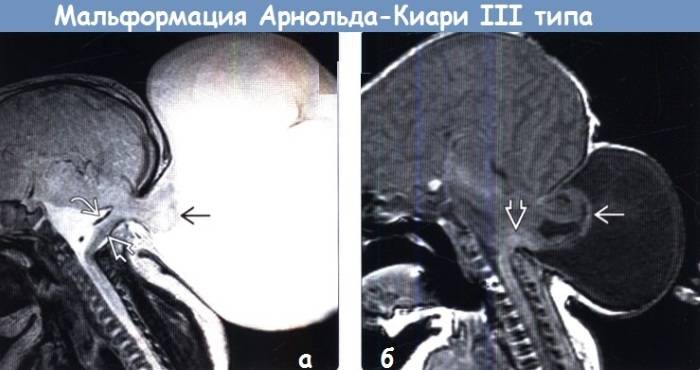
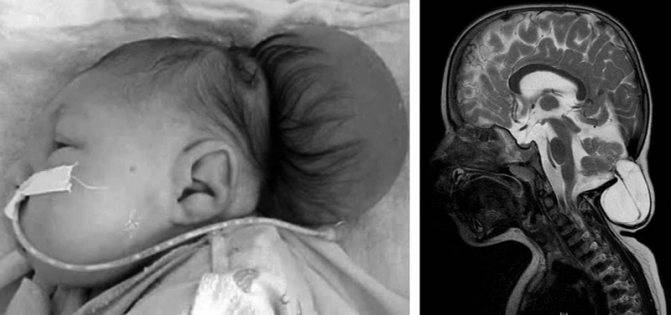
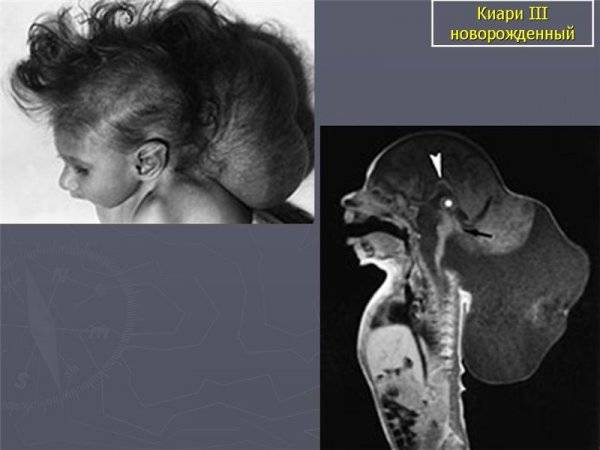
- III тип патологии характеризуется опущением малого и продолговатого мозга до менингоцеле шейно-затылочной области.
- IV тип патологии являет собой недоразвитость мозжечка без опущения миндалин в БЗО.
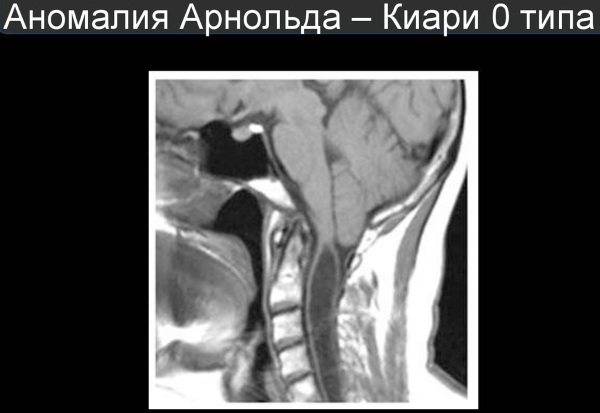
В начале двадцать первого века был выделен нулевой тип аномалии. Его определяет наличие гидромиелии без опущенных миндалин малого мозга.
Внимание! IV тип мальформации Арнольда-Киари стали относить к патологии Денди-Уокера. Еленой Потемкиной, после ряда исследований, было предложено классифицировать синдром по степени опускания миндалин мозжечка в затылочное большое отверстие:
Еленой Потемкиной, после ряда исследований, было предложено классифицировать синдром по степени опускания миндалин мозжечка в затылочное большое отверстие:
- 0 степень – миндалины находятся на верхнем уровне ЗО;
- 1 степень – миндалины сместились до верхнего края дуги атланта или С1;
- 2 степень – на уровне верхней кромки С2;
- 3 степень – на уровне верха С3 и ниже, в каудальном направлении.
Симптоматика
Признаки аномалии Арнольда-Киари могут быть как самостоятельными, так и общими.
К ним относятся следующие симптомы, на которые стоит обратить внимание:
- При повороте головы и в момент покоя в ушах слышится необычный нарастающий звук в виде шума, гула, свиста;
- При физической нагрузке из-за напряжения появляется головная боль;
- Подергивание глазных яблок или нистагма;
- Боль в затылке по утрам. Повышение внутричерепного давления, онемение;
- Из-за нарушения деятельности мозжечка могут наблюдаться головокружения и потеря ориентации;
- В движениях наблюдается неуклюжесть;
- При тяжелой форме болезни появляется тремор конечностей. Что такое тремор конечностей читайте здесь.
- Появляются проблемы со зрением в виде «пелены на глазах», раздвоения предметов, слепоты. При поворотах головы явления усиливаются. Связанно это с давлением на продолговатый мозг;
- Мышцы конечностей, лица и туловища становятся значительно слабее;
- Проблемы с мочеиспусканием наблюдаются при запущенной и тяжелой форме заболевания;
- Порог чувствительности становится меньше в одной из частей тела или лица, конечности;
- При резких поворотах головы больной может потерять сознание;
- Нарушение в работе дыхательной системы – тяжелое дыхание или полная его остановка;
- Тяжелые осложнения – паралич конечностей, гидроцефалия, образование кисты в позвоночнике, деформация костей ступни.
Отличительным признаком синдрома I типа является непрекращающаяся головная боль. При II типе, который проявляется сразу после рождения или в раннем детстве можно наблюдать проблемы с глотанием, слабый крик.
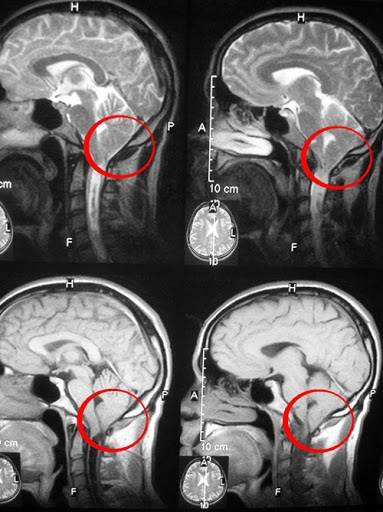
Все перечисленные выше симптомы становятся сильнее при движении.

Внимание! При несвоевременном обращении к врачу и отсутствии лечения может произойти инфаркт головного или спинного мозга
Этиология
Причины синдрома в настоящее время точно не определены. Существует несколько теорий происхождения патологии, но до конца ни одна из них не имеет официального подтверждения. Мнения ученых-неврологов всего мира до сих пор расходятся.
Большинство медиков признают синдром врожденным недугом, сформированным в процессе эмбриогенеза под воздействием негативных факторов внешней среды, оказывающих свое пагубное действие на женский организм при беременности. К ним относятся: самостоятельные использование лекарств, алкоголизм, табакокурение, вирусные инфекции, ионизирующее излучение.
Врожденные причины:
- Дистопия мозжечковых миндалин за пределы задней черепной ямки, имеющей относительно малые размеры;
- Выталкивание растущих структур мозга через затылочное отверстие;
- Неправильное формирование в процессе эмбриогенеза и атипичное развитие в постнатальном периоде костной ткани, приводящее к деформации черепной коробки.
Некоторые ученые определенную роль в развитии синдрома отводят генетическому фактору. В настоящее время можно точно утверждать, что болезнь не связана с хромосомными аномалиями.
Другие ученые придерживаются иной точки зрения относительно происхождения синдрома. Они считают его приобретенным и объясняют свое мнение появлением симптомов патологии у взрослых лиц. Приобретенный синдром возникает под действием экзогенных факторов. У больного новорожденного ребенка череп может иметь нормальное строение без костных аномалий и гипоплазии.
Приобретенные причины:
- Родовой травматизм с поражением черепа и мозга,
- Воздействие цереброспинальной жидкости на стенки спинного мозга,
- Любые ЧМТ,
- Бурный рост мозга в условиях медленно растущих костей черепа.
Симптомы патологии долгое время отсутствуют у больных, а затем внезапно появляются под воздействием провоцирующих факторов: вирусов, травм головы, стрессов.
Опущение основных мозговых структур до шейных позвонков блокирует процесс перетекания ликвора из подпаутинного пространства в спинномозговой канал. Это приводит к дисциркуляторным изменениям. Ликвор, продолжая синтезироваться и никуда не оттекая, скапливается в головном мозге.
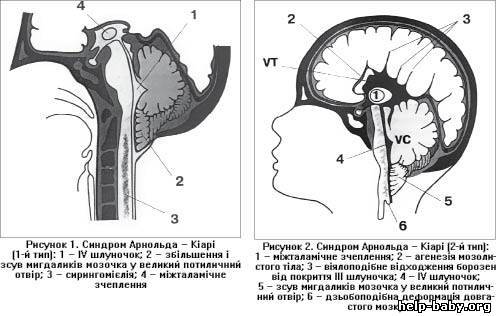
Ученый Киари в 1891 году выделил четыре типа аномалии:
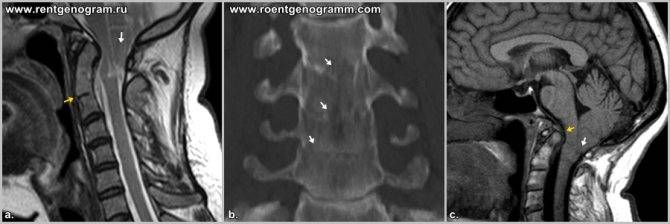
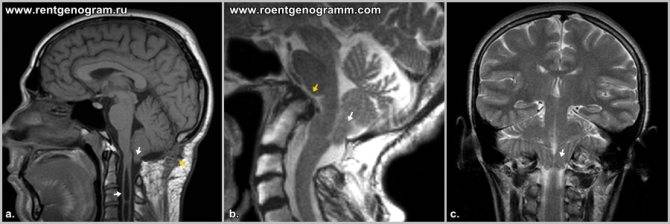
I – выход структурных элементов головного мозга за пределы задней черепной ямки, обусловленный недоразвитием костной ткани этой области. Этот тип клинически проявляется у лиц зрелого возраста.
выход структур мозжечка за пределы ЗЧЯ при аномалия 1 типа
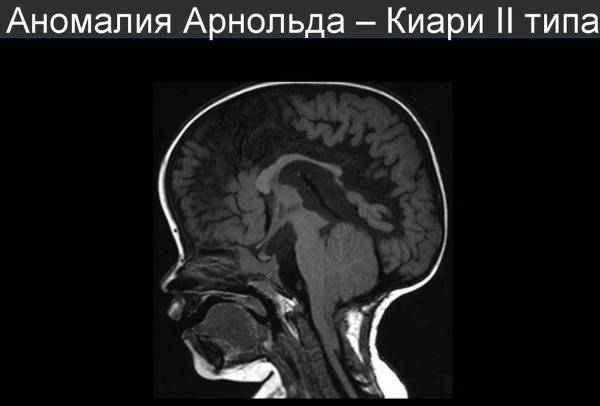
II — нарушения эмбриогенеза, приводящие к расположению структур мозжечка и продолговатого мозга ниже большого затылочного отверстия.
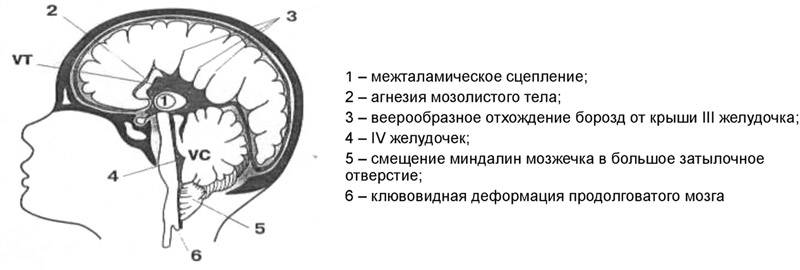
синдром Арнольда-Киари II типа
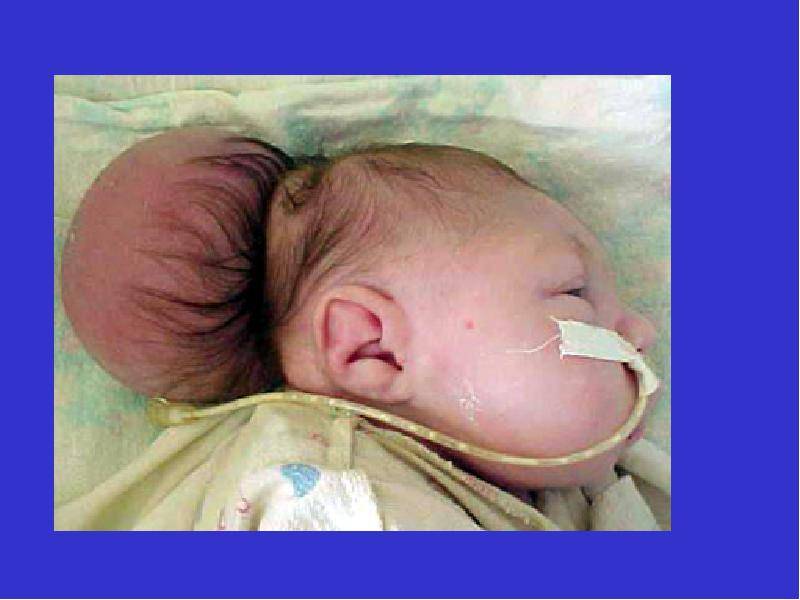
III — эктопия мозговых структур в каудальном направлении с образованием энцефаломенингоцеле.
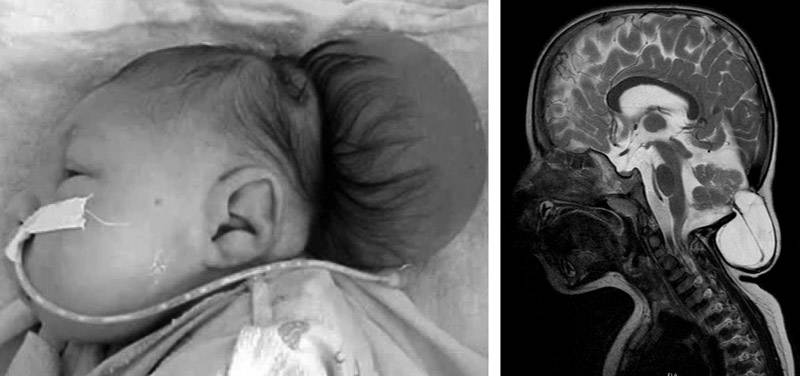
аномалия III типа
IV — недоразвитый мозжечок не смещается и не выходит за пределы черепа. Поскольку отсутствует грыжевое выпячивание мозга, этот тип синдрома отсутствует в современной классификации.

Существуют два новых типа синдрома. Тип — мозжечок располагается достаточно низко, но при этом находится в черепной коробке. Тип 1.5 – промежуточная форма, сочетающая признаки I и II типов.
Выделяют три степени тяжести патологии:
- Первая — относительно легкая форма патологии без аномалий мозговых структур и характерных клинических проявлений.
- Вторая — наличие пороков развития ЦНС с врожденным недоразвитием головного мозга и подкорки.
- Третья — аномалии строения головного мозга со смещением мягких тканей в отношении твердых структур, образованием ликворных кист и сглаженностью извилин.
Этиология мальформации Киари
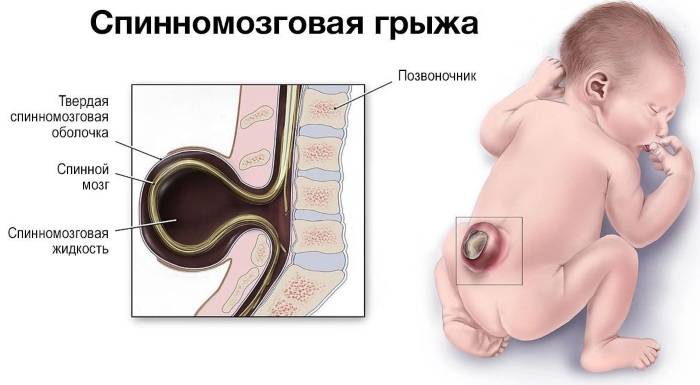
Этиология заболевания в настоящее время не ясна. Имеются данные, свидетельствующие о роли генетического фактора в этиологии этого синдрома. Эктопия миндалин мозжечка в затылочное отверстие была обнаружена у трех монозиготных близнецов. После первого описания мальформации Cleland в 1883 г. появилось несколько теорий. Теория, подтверждаемая исследованиями Misao Nishikawa и соавторов, заключается в том, что из-за парааксиальной дисплазии мезодермального листка или первичного повреждения структур соответствующего сомита формируется ненормально маленькая задняя черепная ямка, структуры заднего мозга, заполнив объем задней черепной ямки и продолжая расти, опускаются в затылочный канал. Сочетание Аномалии Киари II типа с менингомиелоцеле связано с тем, что степень парааксиальной дисплазии мезодермального листка при АК – II типа более выражена, чем при АК – I типа и отмечается не только на уровне формирования затылочной кости, но и по оси тела на уровне формирования ряда позвонков, что проявляется в spina bifida, а также в аномалиях ряда других костных структур и костной системы в целом.
Они помогают точно определить диагноз, а также тип искажения, о котором идет речь. В первые месяцы жизни нельзя различать мозжечки. Кроме того, аномалии нижних черепных нервов могут возникать в нескольких комбинациях. Если пациент выживет до конца детства или подросткового возраста, может возникнуть один из синдромов, которые присутствуют с мальформацией типа Арнольда Киари.
Прогрессирующая мозжечковая атаксия. Сирингомиелии. Пациенты могут присутствовать с сочетанием симптомов нижних черепных нервов, мозжечка, луковицы и спинного мозга, обычно в сочетании с головной болью. Болезнь часто путают с рассеянным склерозом или с опухолью малютки или нижнего шейного шнура. Симптомы могут иметь острое начало после расширения, хиропрактики или удаления зубов. Физическая привычка таких пациентов может быть нормальной, но около 25% имеют признаки того, что гидроцефалия остановлена короткой шеей.
У детей
Чаще всего отмечается блокада внутри желудочковой системы и представляет собой несообщающуюся гидроцефалию. Это обычно вызвано сужением сильвиева водопровода либо недоразвитием отверстий Мажанди и Люшки. Как генерализованное нарушение развития нервной системы, оно может сочетаться с микро- или макрогирией, порэнцефалией, агенезией мозолистого тела, слиянием полушарий мозга, агенезией червя мозжечка, позвоночных расщелин, менингоцеле, энцефалоцеле, сирингомиелией, гидромиелией. Причиной сообщающейся гидроцефалии может быть процесс образования спаек в субарахноидальной области в основания мозга, перинатальные субарахноидальные или внутрижелудочковые кровоизлияния.
Синдром у детей может возникать в результате травматических повреждений клиновидно-затылочной и клиновидно-решетчатой области при родовых травмах. Клинические проявления сводятся к нарушению кормления (рефлюксу и аспирации), эпизодам апноэ, стридору, нарушениям краниальной иннервации, судорожным приступам устойчивым к противосудорожным препаратам.
Синдром Арнольда Киари у плода
Патология Киари является врожденным остеоневропатией и формируется на этапе закладки и развития нервной трубки у плода, асинхронным формированием нервного ствола и костей черепной ямки.
Морфологические проявления данной патологии можно выявить во время проведения ультразвукового исследования, предопределить дальнейший прогноз. Чаще всего выявление патологии становится причиной для прерывания беременности.
Другие признаки
Они наблюдаются при более тяжёлой степени синдрома. Первоисточниками инфаркта головного или спинного мозга часто бывают эти признаки аномалии Арнольда Киари: 1 тип не такой страшный, как его другие подвиды. Для них характерно не только двоение в глазах, но и полная слепота, потеря сознания, нарушение координации, тремор конечностей, проблемы с мочеиспусканием, слабость мышц, потеря чувствительности большой части тела (иногда целой половины туловища).
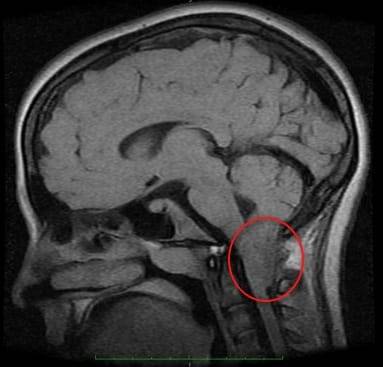
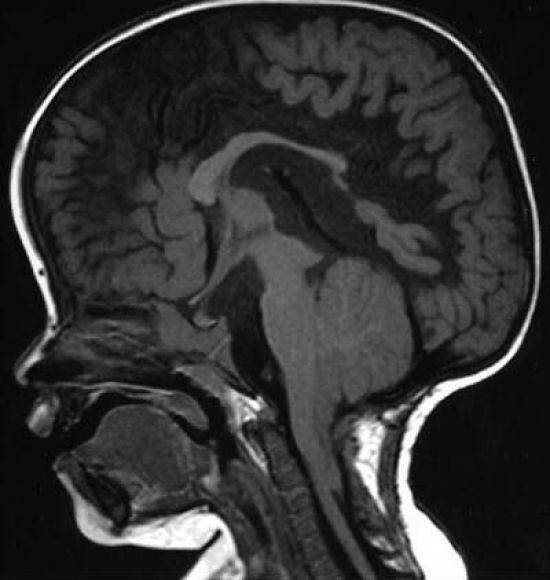
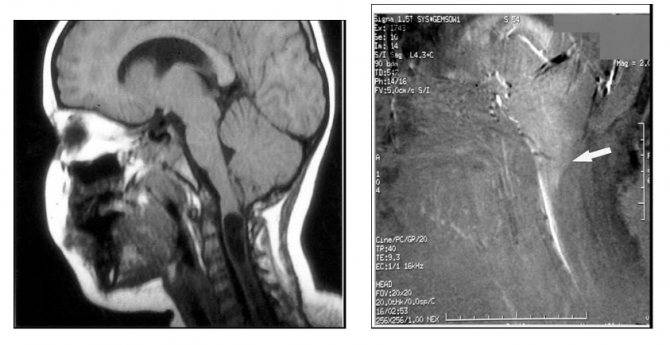
Развитие разных последствий для здоровья может спровоцировать аномалия Арнольда Киари 1 типа. Продолжительность жизни и её нормальное течение зависят от быстрой и точной диагностики. Единственным методом обнаружить патологию является МРТ (магнитно-резонансная томография) мозга. Она требует от пациента состояния полной неподвижности, поэтому активных младенцев усыпляют специальными медикаментами.
Болезнь сопровождается специфическими синдромами. Выраженность и количество этих симптомокомплексов зависит от тяжести порока, какие внутричерепные и позвоночные структуры затронуты патологией.
| Название синдрома | Характер проявления | Симптомы аномалии |
|---|---|---|
| Церебральный (мозжечковый) | Расстройство координации движений, неврологические нарушения, неконтролируемый нистагм (непроизвольное подергивание, колебание глаз) | Нет точности мелкой моторики, походка шаткая, неустойчивость вертикального положения тела.
Тремор (дрожание) пальцев, ухудшение речи, двоение картинки (нарушение зрения). У детей отмечается умственная недостаточность |
| Гипертензионно-гидроцефальный | Нарушение оттока и всасывания ликвора, повышенное внутричерепное давление (интракраниальная гипертензия), водянка мозга | Распирающая головная боль.
Тошнота, рвота вне зависимости от приема лекарств, пищи, напряжение мышц на затылке. Ухудшение самочувствия после пробуждения, движения головой. |
| Бульбарно-пирамидный | Сдавливание пирамид, моста, продолговатого мозга | Паралич либо парез (слабость) мышечной группы на руке, туловище.
Боль/дискомфорт в зоне затылка. Усиливается во время чихания, движений головы. 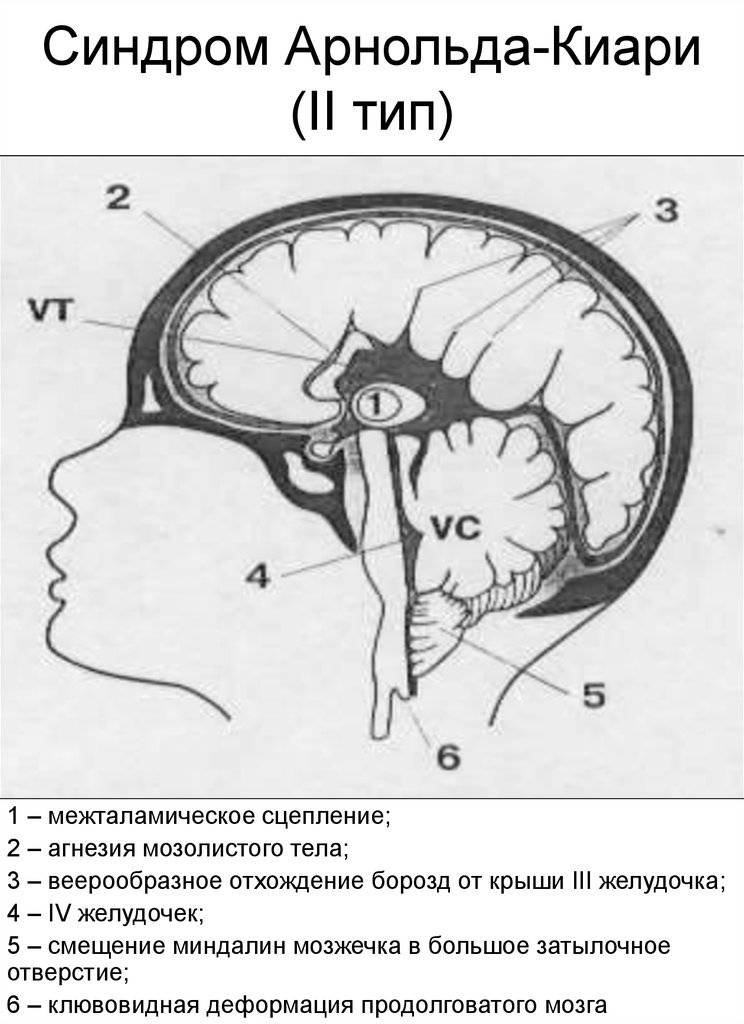

Онемение участков кожи, тела. Нарушение глотательной, дыхательной, речевой функции. |
| Вертебробазилярной недостаточности | Сдавливание кровеносных сосудов, ухудшение питания мозга, кислородное голодание тканей (гипоксия) | Приступы головокружения, полуобморочное состояние, потери сознания.
Ухудшение мышечной силы. Нарушения зрения, слуха. |
| Корешковый | Поражение подъязычного, блуждающего, других нервов и ядер в зоне затылочной кости, большого отверстия, атланто-аксиального сустава, мозжечка | Онемение в области лица, языка.
Нарушения глотания, речи, слуха. |
| Сирингомиелитический | Развитие кисты в веществе продолговатого/спинного мозга, сохраняется восприятие тела в пространстве | Пропадает на коже предплечий, рук, туловища чувствительность к раздражителям, смене температуры (нет боли от ожогов, ран, обморожения).
Слабость и атрофия мышц конечностей, туловища, головы, деформация мелких суставов, искривление позвоночника. Возможно недержание кала, мочи, иная дисфункция выделительных органов. |
Наблюдаются признаки скачков давления и гипоксии:
- головокружение;
- перед глазами мелькание «мушек»;
- шум в ушах;
- слабость.
Человек ощущает дискомфорт сзади у основания черепа, при поворотах/наклонах головы. В случае прогрессирования возможно присоединение других симптомокомплексов. Это проявление бульбарно-пирамидного, корешкового и/или сирингомиелического синдрома.
Признаки 2 степени
Пороку Арнольда-Киари 2 типа характерно:
- систематическая остановка дыхания;
- ослабление глоточных мышц;
- синюшность кожи.
Осложняется вторая степень частичным либо полным параличом. Утрата двигательных функций отмечается в пальцах, руке/ноге, в области головы, всего тела.
Признаки 3 степени
Третьей степени мальформации Арнольда-Киари характерно проявление 5―6 синдромов одновременно. Ребенок не кричит сразу после рождения. У пациентов часто отмечается потеря слуха, зрения, дисфункция выделительных органов. Нет четкой координации движения, по утрам возникает рвота, есть аномалии строения позвоночника, головы.
Признаки 4 типа
У людей с четвертой степенью тяжести порока преобладает мозжечковый и гипертензионно-гидроцефальный синдром. Дети беспокойны, слабо вскрикивают, тихо плачут. Если выживают, на обследованиях выявляют снижение интеллекта, отсутствие нормального физического развития. В целом, симптомы с прогнозом жизни третьей и четвертой степени тяжести похожи.
Проявления синдрома Арнольда-Киари
Симптоматика синдрома Арнольда-Киари определяется его типом и характером смещения структур ЗЧЯ. Часто он протекает бессимптомно и обнаруживается случайно в ходе обследований мозга. У взрослых появление симптоматики может спровоцировать травма головы, у малышей некоторые формы заболевания заметны уже в первые часы и дни жизни.
Аномалия I типа диагностируется наиболее часто и может проявиться в подростковом либо взрослом возрасте следующими синдромами:
- Гипертензионным;
- Церебеллярным;
- Бульбарным;
- Сирингомиелическим;
- Явлениями поражения черепных нервов.
Гипертензионный синдром вызван увеличением внутричерепного давления вследствие блокады оттока ликвора сместившимися отделами мозга. Он проявляется:
- Головными болями в области затылка, особенно, при чихании, кашлевых толчках;
- Тошнотой и рвотой, после которой больной не ощущает облегчения;
- Напряжением мышц шеи.
Признаками вовлечения мозжечка (церебеллярный синдром) считают расстройства речи, двигательной функции, равновесия, нистагм. Пациенты жалуются на шаткость походки, неустойчивость положения тела в пространстве, затруднение мелкой моторики и четкости движений.

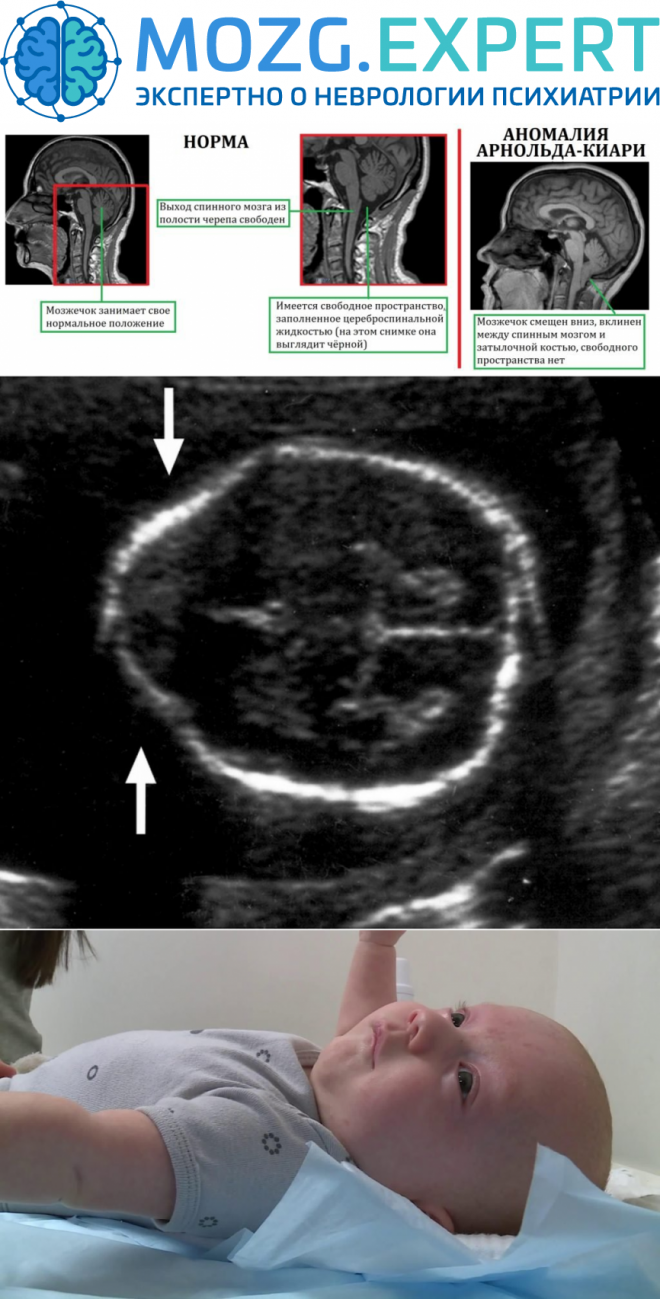

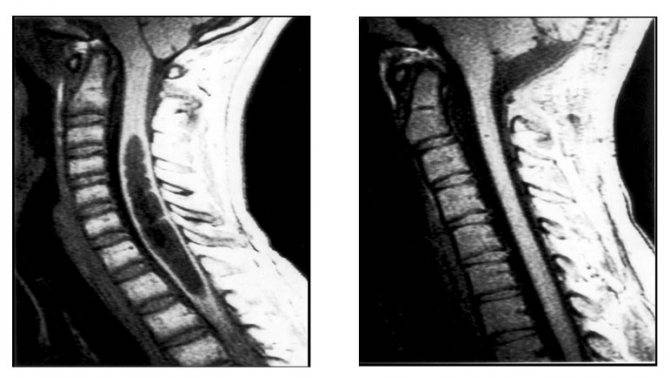
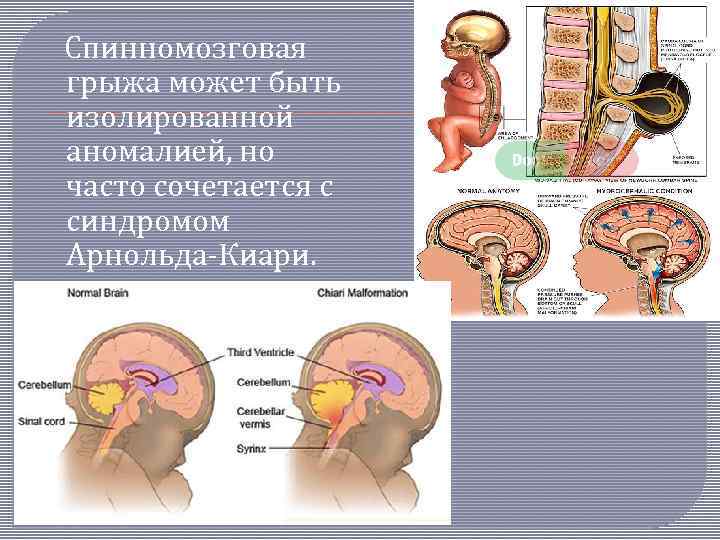
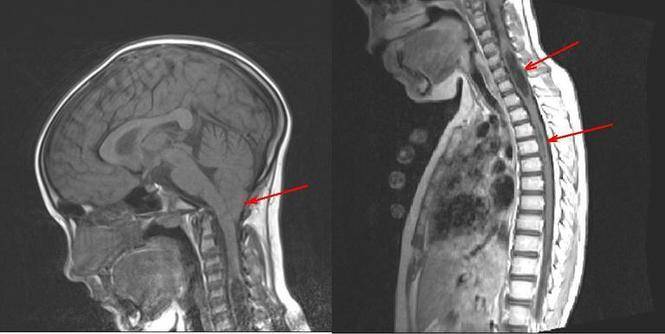

Поражение стволового отдела мозга представляется опасным ввиду расположения там ядер черепных нервов и жизненно важных нервных центров. Стволовая симптоматика состоит в:
- головокружении; двоении в глазах и снижении зрения;
- затруднении глотания;
- ухудшении слуха, шуме в ушах;
- обмороках, гипотонии, ночных апноэ.
При образовании полостей и ликворных спинномозговых кист на фоне затрудненного тока спинномозговой жидкости у пациентов с I вариантом мальформации возникают признаки сирингомиелического синдрома — расстройство чувствительной сферы, онемение кожи, гипотрофия мышц, дисфункция тазовых органов, снижение и исчезновение брюшных рефлексов, периферические нейропатии и изменения со стороны суставов.
Расстройства чувствительности сопровождаются нарушением восприятия собственного тела, когда пациент, закрыв глаза, не может сказать, в каком положении находятся его руки или ноги. Снижается также чувствительность к боли и температуре.
По наблюдениям неврологов, диаметр и локализация кисты спинного мозга не обязательно отражаются на выраженности и распространенности нарушений чувствительной и двигательной сферы, гипотрофии мышц.
При синдроме второго и третьего типа течение патологии значительно тяжелее, симптоматика появляется у ребенка сразу же после родов. Характерны нарушения дыхания — стридор (шумное дыхание), приступы его остановки, а также двусторонний парез гортани, провоцирующий расстройства глотания, когда жидкая еда попадает в носовые ходы.
Второй тип аномалии у малышей первых месяцев жизни сопровождается нистагмом, усилением тонуса мышц в руках, синюшностью кожи, которые особенно заметны при кормлении грудничка. Двигательные нарушения вариабельны, проявления их меняются, возможна тетраплегия — паралич и верхних, и нижних конечностей.
Мальформация Арнольда-Киари может приводить к осложнениям, вызванным блокадой тока ликвора, поражением ядер черепных нервов, ущемлением стволовых структур. Самыми частыми считаются:
- Гипертензионно-гидроцефальный синдром — увеличение внутричерепного давления вследствие блокады путей ликворооттока, возможен как у детей, так и у взрослых;
- Расстройства дыхания, апноэ;
- Инфекционно-воспалительные процессы — бронхопневмонии, уроинфекции, которые связаны с лежачим положением пациента, нарушением актов глотания и дыхания, функции тазовых органов.
При тяжелом течении патологии может наступить кома, остановка сердца и дыхания, которые приводят к смерти в считанные минуты. Реанимационные мероприятия позволяют обеспечить витальные функции, но вернуть к жизни мозг и устранить необратимые последствия компрессии его отделов, к сожалению, практически невозможно.
Симптомы
Неспецифические признаки, на которые стоит обратить внимание: неустойчивая, неровная походка, сбои в работе вестибулярного аппарата, что выражается в нарушении равновесия, расстройство мелкой моторики кистей рук. Основные симптомы, указывающие на наличие аномалии Арнольда-Киари:
- Боль в зоне шеи и затылка, которая становится более интенсивной при кашле, внезапных, быстрых поворотах головы, физическом перенапряжении.
- Гул, звон, шум в ушах.
- Ухудшение кожной чувствительности к боли и изменению температуры в зоне конечностей.
- Мышечная слабость в области верхних конечностей.
- Спастика, судорожный синдром, проявляющийся в конечностях.
- Онемение конечностей.
- Головокружения, нередко приводящие к обмороку.
- Зрительная дисфункция – диплопия (двоение предметов в глазах), анизокория (разный диаметр зрачков), светобоязнь, болезненные ощущения при движении глазными яблоками, спазм аккомодации.
Головная боль, локализующаяся в шейно-затылочном отделе и усиливающаяся при физическом перенапряжении или чихании – основной и самый первый характерный симптом, который выявляется у 75% пациентов. Нарушения кожной чувствительности встречаются в 60% случаев с локализацией преимущественно в области верхних конечностей. Гемипарезы (частичный паралич в одной стороне тела) наблюдаются у 40% больных.
Прогрессирование, усугубление болезни Арнольда-Киари вызывает дополнительные симптомы – апноэ (кратковременная остановка дыхания), ослабление глотательного рефлекса, дисфагия (неприятные, болезненные ощущения при глотании), нистагм (непроизвольные частые движения глазных яблок). Скандированная речь и атаксия (нарушение согласованности при сокращении разных групп мышц) появляются вследствие компрессии тканей мозжечка.
Симптомы, характерные для аномалии Арнольда-Киари в статусе 1 или 2 типа, свойственны многим заболеваниям – рассеянному склерозу, опухоли мозга, шейному остеохондрозу, позвоночной грыже, карпальному синдрому (поражение нервных окончаний кисти рук, что приводит к болезненным ощущениям и онемению). Для дифференцирования патологии проводится развернутое диагностическое обследование.
Симптоматика

Аномалия Киари I типа — самая распространенная форма синдрома, клинические признаки которой условно объединены в пять синдромов:
- Гипертензионный синдром проявляется цефалгией, подъемом артериального давления в утренние часы, напряженностью и гипертонусом шейных мышц, дискомфортом и болезненными ощущениями в шейном отделе позвоночника, диспепсическими явлениями, общей астенизацией организма У новорожденных детей возникает общее беспокойство, рвота фонтаном, тремор подбородка и конечностей, нарушается сон. Ребенок постоянно плачет, отказывается от груди.
- При наличии мозжечковых нарушений у больных изменяется произношение, речь становится скандированной, возникает вертикальный нистагм. Они жалуются на частые головокружения, рассогласованность движений, шаткость походки, дрожание рук, нарушение равновесия, дезориентацию в пространстве. Больные с большим трудом выполняют простые целенаправленные действия, в движениях отсутствует четкость и скоординированность.
- Поражение черепно-мозговых нервов проявляется признаками корешкового синдрома. У пациентов ограничивается подвижность языка и мягкого неба, что приводит к нарушению речи и проглатывания пищи. Их голос изменяется в сторону гнусавости и осиплости, речь становится неясной, дыхание затрудненным. Нарушение ночного дыхания отмечаются у большинства больных. У них возникает гипопноэ, центральное или обструктивное апное, при прогрессировании которого развивается острая дыхательная недостаточность. Лица с синдромом плохо слышат и видят, у них двоится в глазах и шумит в ушах. Со стороны органов зрения пациенты отмечают наличие светобоязни и боль при движении глазными яблоками. Офтальмологи часто обнаруживают анизокорию, спазм аккомодации или скотомы. Одним из основных симптомов синдрома является гипестезия – снижение чувствительности кожи лица и конечностей. Подобные патологическое изменения связаны с приглушенным реагированием рецепторов кожи на внешние раздражители: тепло или холод, уколы, удары. В тяжелых случаях нервное окончания вообще перестают воспринимать различные экзогенные воздействия.
- Сирингомиелический синдром — сложный симптомокомплекс, проявляющийся парестезией или онемением конечностей; изменением тонуса мышц и их гипотрофией, приводящей к миастеническим расстройствам; поражением периферических нервов, проявляющимся болью в конечностях; дисфункцией органов таза в виде затрудненной дефекации или самопроизвольного мочеиспускания; возможны артропатии — поражения суставов.
- У больных с пирамидальной недостаточностью снижается сила в нижних конечностях и способность к тонким движениям, ограничивается объем движений, повышается мышечный тонус — так называемая спастичность, например, спастическая походка. Повышение сухожильных рефлексов сочетается с одновременным снижением кожных рефлексов — брюшных. Возможно появление патологических рефлексов. У пациентов страдает мелкая моторика рук.
Любое неосторожное движение усиливает симптомы патологии, делает их более выраженными и яркими. Изменение положения головы — частая причина потери сознания
Профилактика
Поскольку этиология синдрома окончательно не выяснена и нет конкретной информации о его патогенезе, предупредить развитие патологии не представляется возможным. Будущим родителям необходимо знать все о ведении здорового образа жизни и при планировании беременности стараться соблюдать указанные правила:
- Отказаться от пагубных привычек в виде табакокурения и употребления спиртных напитков,
- Обогащать свой рацион белковыми продуктами, фруктами, овощами, ягодами, исключив из него сладости и вредности,
- Своевременно обращаться к врачам за медицинской помощью,
- Принимать лекарственных препараты по назначению врача и в строго указанных дозировках,
- С профилактической целью принимать поливитамины,
- Беречь свое здоровье и наслаждаться жизнью.